Активация митохондриального окисления путем ингибирования
PDK2 обращает вспять устойчивость к цисплатину при раке головы и шеи
Оригинал статьи: https://www.sci-hub.ru/10.1016/j.canlet.2015.11.023
Чон Лиел Рох a, *, Джин Янг Парк a, Ын Хе Ким a, Хе Джин Чан a, Минсу Квон b
a Отделение отоларингологии, Медицинский центр Асан, Медицинский колледж Университета Ульсан, Сеул, Республика Корея
b Отделение отоларингологии, Медицинская школа при больнице Национального университета Кёнсан, Чинджу, Республика Корея
Автор переписки: Тел: +82 2 3010 3965; факс: +82 2 489 2773. Адрес электронной почты: rohjl@amc.seoul.kr (J.-L. Roh).
Получено: 9 сентября 2015 г.
Пересмотрено: исправленная форма 13 ноября 2015 г.
Принято: 14 ноября 2015 г
Аннотация
Дихлорацетат (ДХА), сиротский препарат, способствующий переходу от гликолиза к окислительному фосфорилированию, был повторно использован для лечения рака. В настоящем исследовании изучалось, может ли DCA преодолеть резистентность к цисплатину при раке головы и шеи (РГШ). Были использованы две устойчивые к цисплатину линии клеток РГШ (AMC-HN4R и -HN9R), их родительские линии и другие линии клеток РГШ человека. Эффект DCA, отдельно и в комбинации с цисплатином, оценивали путем измерения клеточного цикла, жизнеспособности, гибели, продукции реактивных форм кислорода (ROS), мембранного потенциала митохондрий (ΔΨm) и экспрессии белков в доклинических моделях опухолевых ксенотрансплантатов мыши. Увеличение гликолиза коррелировало со снижением чувствительности к цисплатину и уменьшалось под действием DCA. Устойчивые к цисплатину клетки сверхэкспрессировали киназу пируватдегидрогеназы 2 (PDK2). DCA индуцировал гибель клеток HNC путем снижения ΔΨm и стимулирования производства митохондриальной ROS. Этот эффект снижался антиоксидантом N-ацетил-л-цистеином или ингибированием апоптоза, опосредованного каспазой. Активация митохондриального окисления глюкозы под действием DCA в конечном итоге активировала нижележащие митохондриальные апоптотические сигналы, что привело к гибели химиорезистентных раковых клеток. Таким образом, DCA значительно сенсибилизировал устойчивые клетки HNC к цисплатину in vitro и in vivo. Высокий гликолиз и сверхэкспрессия PDK2 тесно связаны с устойчивостью клеток НЯК к цисплатину; последняя может быть преодолена с помощью DCA.
Ключевые слова: Рак головы и шеи, устойчивость к цисплатину, PDK2, дихлорацетат, ремоделирование митохондрий
Сокращения: РГШ, рак головы и шеи; ДХА, дихлорацетат; CDDP, цисплатин; OXPHOS, окислительное фосфорилирование; PDK2, киназа 2 пируватдегидрогеназы; PDHE1α, изоформа E1α пируватдегидрогеназы; ROS, реактивные виды кислорода; ΔΨm, мембранный потенциал митохондрий; NAC, N-ацетил-цистеин; DCF-DA, 2′,7′- дихлорфлуоресцеин диацетат; PARP, поли(АДФ-рибоза) полимераза; siRNA, короткая интерферирующая РНК; 18F-FDG, 18F-фтордезоксиглюкоза; PET, позитронно-эмиссионная томография; SUV, стандартизированное значение поглощения; MTV, метаболический объем опухоли; TUNEL, терминальная дезоксинуклеотидилтрансфераза-опосредованное мечение ник-конца ДУТФ.
© 2015 Elsevier Ireland Ltd. Все права защищены.
ВВЕДЕНИЕ
Рак головы и шеи (РГШ) является восьмым по распространенности раком во всем мире, ежегодно диагностируется более полумиллиона новых случаев [1]. Опухоли возникают в верхних отделах пищеварительного тракта, включая ротовую или носовую полость, глотку и гортань, и метастазируют в регионарные лимфатические узлы и отдаленные участки. Современный стандарт лечения ГНК включает мультимодальный подход, включающий хирургию, химиотерапию и радиотерапию, особенно при распространенном ГНК. Наряду с недавним интересом к стратегиям сохранения органов, нехирургические методы, такие как радиотерапия в сочетании с химиотерапией, все чаще используются в качестве первой линии терапии при НЯК [2]. В настоящее время системная химиотерапия является центральным компонентом нескольких лечебных подходов, включая комбинацию с окончательной лучевой терапией или индукционным лечением, превышающим резервирование только для паллиации [3]. Цисплатин, платиновое производное цис-диамминдихлорплатина (II) (CDDP), остается химиотерапевтическим препаратом первой линии в нехирургических методах лечения НЯК, несмотря на последние достижения в области таргетной терапии [4]. Однако за последние три десятилетия, несмотря на диагностические и терапевтические достижения, общая выживаемость при НЯК существенно не изменилась, что является результатом стойкой резистентности раковых клеток к терапии, включая цисплатин и облучение [2].
Метаболические изменения являются общей чертой раковых клеток: сдвиг в генерации клеточной энергии от митохондриального окислительного фосфорилирования к аэробному гликолизу обеспечивает биосинтетическое преимущество раковых клеток [5]. Увеличение гликолиза в раковых клетках, называемое эффектом Варбурга, обычно связано с фенотипическими изменениями, включая адаптацию к гипоксии и недостатку питательных веществ, устойчивость к окислительному стрессу и апоптотическим стимулам, а также повышенный синтез биомассы [6]. Дерегулированный метаболизм связан с устойчивостью к лечению при терапии рака [7]. В гликолитическом пути повышение уровня транспортера глюкозы (GLUT), гексокиназы (HK), пируваткиназы M2 (PKM2), киназы пируватдегидрогеназы (PDK), лактатдегидрогеназы-A (LDHA), синтеза жирных кислот (FASN) и глутаминазы, среди прочих, связано с устойчивостью к противораковым препаратам [7]. Имеются данные о том, что регулирование метаболизма раковых клеток может улучшить терапию и преодолеть устойчивость к химиотерапии или радиотерапии [8,9].
Дихлорацетат (ДХА), сиротский препарат для лечения молочнокислого ацидоза, сдвигает метаболизм раковых клеток с гликолиза на окислительное фосфорилирование [10]. DCA избирательно ингибирует PDK, который активируется во многих видах рака, что приводит к активации пируватдегидрогеназы (PDH), комплекса ферментов, которые превращают цитозольный пируват в митохондриальный ацетил-КоА. Ингибирование PDK с помощью малой интерферирующей РНК (siRNA) или лечение DCA изменяет биоэнергетику рака и восстанавливает митохондрий-зависимый апоптоз в раковых клетках [11]. DCA эффективен в лечении рака с агрессивным фенотипом in vitro и in vivo[12,13], а также преодолевает резистентность к сорафенибу в гепатоцеллюлярной карциноме путем активации митохондриального окислительного фосфорилирования [14]. Таким образом, DCA может быть подходящим противораковым препаратом для повышения эффективности химиотерапии и преодоления резистентности к химиотерапевтическим средствам при раке человека; однако, хотя DCA широко изучался при раке, он редко тестировался на клетках НЯК [15], особенно в условиях резистентности к химиотерапевтическим средствам. Необходимо дальнейшее изучение механизмов действия DCA и синергизма с традиционными химиотерапевтическими препаратами. Здесь мы показываем, что DCA смещает биоэнергетику НЯК в сторону митохондриального окисления глюкозы и приводит к накоплению в клетках митохондриальных реактивных форм кислорода (mROS), тем самым сенсибилизируя химиорезистентные клетки НЯК к цисплатину in vitro и in vivo.
Материалы и методы
Культура клеток и создание цисплатин-устойчивых клеток НГШ
Клетки рака головы и шеи человека (РГШ) AMC-HN2, -HN3, -HN4, -HN5, -HN9 и -HN10 выращивали в минимальной основной среде Орла (Life TechnologiesTM, Carlsbad, CA, USA); Клетки SNU-1041, -1066 и -1076 выращивали в среде Roswell Park Memorial Institute (Life TechnologiesTM ); а клетки UMSCC1 выращивали в модифицированной среде Дульбекко Eagle (Life TechnologiesTM ). Среда была дополнена 10% фетальной бычьей сывороткой. Все линии раковых клеток были подтверждены путем профилирования ДНК (короткие тандемные повторы, STR), предоставленной Корейским банком клеточных линий. Клетки инкубировали при 37 °C в увлажненной атмосфере, содержащей 5% CO2.
Устойчивые к цисплатину клетки AMC-HN4 и HN9 (HN4R и HN9R) были получены из родительских чувствительных к цисплатину клеток AMC-HN4 и HN9, соответственно, путем воздействия возрастающих концентраций цисплатина (цис-платина (II) диамин дихлорид [CDDP]; Sigma-Aldrich, Луис, МО, США) [16]. Устойчивость к цисплатину в созданных клеточных линиях оценивали с помощью анализа жизнеспособности клеток и сравнивали с родительскими клетками.
Анализ клеточного цикла и гибели клеток
Для анализа клеточного цикла клетки подвергали воздействию ДКА в течение 72 ч. Затем клетки трипсинизировали, фиксировали на ночь в ледяном этаноле и окрашивали в течение 30 мин йодистым пропидием (Sigma-Aldrich) при температуре 37 °С. Содержание клеточной ДНК измеряли с помощью проточного цитометра FACScalibur (BD Bioscience, Сан-Хосе, Калифорния, США). Для анализа клеточной гибели клетки культивировали с цисплатином и DCA, отдельно или в комбинации, или эквивалентным количеством ДМСО (транспортный контроль). Через 72 часа клетки собирали, промывали ледяным PBS и ресуспендировали в буфере для связывания. Клетки окрашивали аннексином V-FITC (флуоресцеин изотиоцианат) и йодистым пропидием с использованием набора для выявления апоптоза аннексином V-FITC (BD Biosciences, Franklin Lakes, NJ, USA), а затем анализировали методом проточной цитометрии. Все данные анализировали с помощью программного обеспечения Cell Quest (BD Biosciences). Статистическую значимость между различными группами лечения оценивали с помощью двуххвостового U-теста Манна-Уитни или t-теста Стьюдента.
Для анализа активности каспазы клетки HN4R и HN9R, посеянные в 96-луночный планшет, подвергали воздействию 100 мкл среды, содержащей цисплатин и DCA отдельно или в комбинации, в течение 72 ч, с или без 3 мМ NAC или 50 мкМ Z-VAD-fmk (R&D Systems, Minneapolis, MN, USA) перед воздействием 30 мМ DCA. Анализы проводили в трех лунках с использованием флуориметрического Homogeneous Caspase Assay (Roche Life Science, Базель, Швейцария). Добавляли рабочий раствор субстрата, и планшет инкубировали в темноте при 37 °C в течение 2-8 ч или при комнатной температуре в течение ночи. Поглощение в каждой лунке измеряли при длине волны возбуждения 485 нм и длине волны эмиссии 520 нм с помощью микропланшетного ридера SpectraMax M2.
Для измерения мембранного потенциала митохондрий (ΔΨm) клетки HN4R и HN9R, посеянные в 96-луночный планшет, подвергались воздействию 100 мкл среды, содержащей цисплатин и DCA отдельно или в комбинации в течение 36 ч. Клетки окрашивались 200 нМ тетраметилродамин этилового эфира (TMRE, Life Technologies TM ) в течение 20 мин и затем анализировались методом проточной цитометрии. Средняя интенсивность флуоресценции (MFI) каждой группы лечения была нормализована по отношению к контрольной группе.
Доклинические исследования
Все процедуры исследования на животных проводились в соответствии с протоколами, утвержденными Комитетом по уходу и использованию животных нашего учреждения. Шестинедельные атимичные самцы мышей BALB/c (nu/nu) были приобретены у Central Lab Animal Inc. (Сеул, Республика Корея). Клетки AMC-HN4R или HN9R (5 x 10 6 ) вводили подкожно во фланг. Объем опухоли и вес тела измеряли каждые 3 дня. Опухоли измеряли с помощью штангенциркуля, а объем рассчитывали как (длина х ширина2 )/2. Лечение начиналось, когда клеточные имплантаты становились пальпируемыми узелками (= день 0). Мыши были рандомизированы на четыре группы лечения: транспортное средство, DCA, цисплатин и DCA плюс цисплатин.
Мышей лечили питьевой водой с добавлением 0,5 г/л DCA, или внутривенным введением 5 мг/кг цисплатина один раз в неделю, или комбинацией DCA и цисплатина по одинаковым графикам. Мышей приносили в жертву на 24-й день, опухоли выделяли и анализировали с помощью иммуноблотинга и in situ терминальной дезоксинуклеотидилтрансферазы-опосредованного мечения никкеля ДУТФ (TUNEL) (EMD Millipore, Billerica, MA, USA). Количество апоптотических телец подсчитывали вслепую в десяти случайно выбранных полях высокой мощности. Статистическую значимость между различными группами лечения оценивали с помощью двуххвостового U-теста Манна-Уитни или t-теста Стьюдента.
Результаты
Увеличение гликолиза в клетках HNC связано с резистентностью к цисплатину и обратимо под действием DCA
Все клеточные линии, использованные в нашем исследовании, были человеческими HNC. Цитотоксический эффект DCA оценивался в клетках HNC с помощью исключения трипанового синего, окрашивания кристаллическим фиолетовым и МТТ. DCA, протестированный в концентрации до 30 мМ в течение 72 ч, заметно подавлял рост клеток HNC (МТТ-тест, рис. 1А). Биоэнергетика клеток НЯК изменялась под действием ДКА: клеточный гликолиз значительно уменьшался, а окислительное фосфорилирование увеличивалось (рис. 1В). Изменение биоэнергетики варьировалось между линиями клеток НЯК и было значительно связано с чувствительностью к цисплатину: клетки НЯК с высоким гликолизом демонстрировали устойчивость к цисплатину (R2 = 0,859, P < 0,001; рис. 1С ). Кроме того, при воздействии DCA раковые клетки с наибольшим изменением биоэнергетики, скорее всего, становились более апоптотичными (R2 = 0,769, P = 0,002; рис. 1D ). Это позволяет предположить, что DCA вызывает специфический для раковых клеток апоптоз путем снижения гликолиза в клетках HNC.
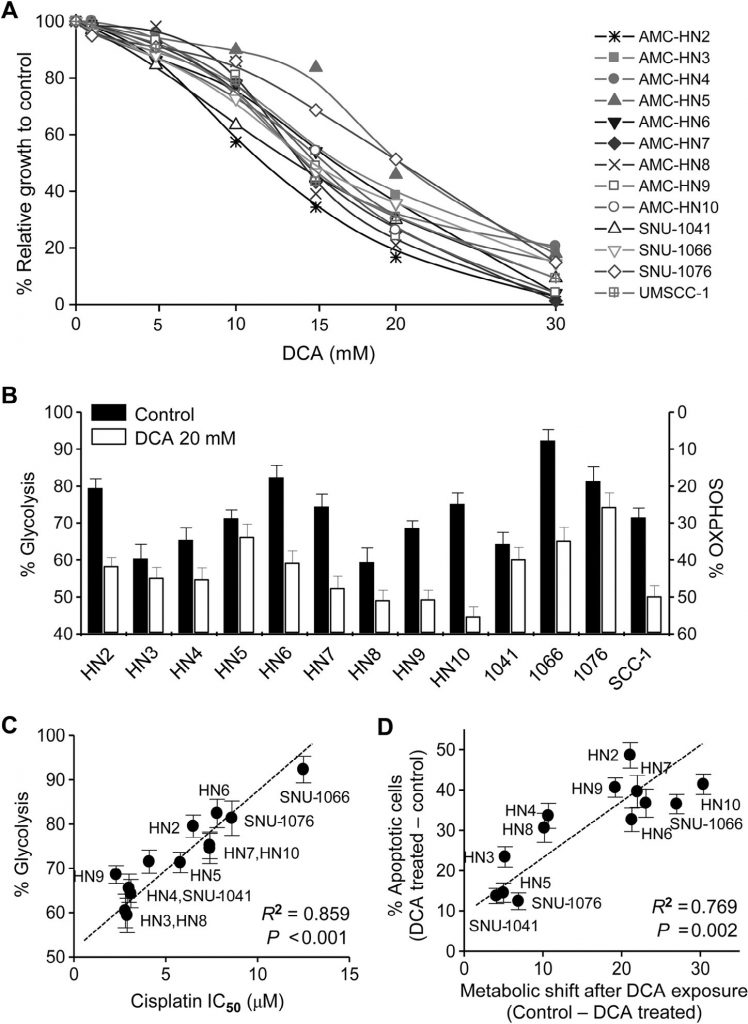 Рис. 1. Повышенный гликолиз в клетках HNC коррелирует со снижением чувствительности к цисплатину и уменьшается под действием дихлорацетата (DCA). (А) DCA индуцировал гибель клеток в клетках HNC. Цитотоксичность оценивалась с помощью МТТ-анализа после воздействия различных концентраций ДХА в течение 72 ч. (B) Изменение биоэнергетики под воздействием ДХА в клетках HNC. Измерения гликолиза и OXPHOS (%) были описаны в разделе «Материалы и методы». (C) Корреляция между гликолизом и IC50 цисплатина в группе клеточных линий HNC. IC50 рассчитывали с помощью МТТ-анализа в трех независимых экспериментах, каждый из которых проводился с использованием трех экземпляров образцов. Корреляция оценивалась с помощью линейной регрессии. (D) Корреляция между биоэнергетическими изменениями и апоптозом в клетках HNC. В анализе апоптоза измеряли аннексин V-положительные апоптотические фракции после воздействия 20 мМ DCA в течение 72 часов.
Рис. 1. Повышенный гликолиз в клетках HNC коррелирует со снижением чувствительности к цисплатину и уменьшается под действием дихлорацетата (DCA). (А) DCA индуцировал гибель клеток в клетках HNC. Цитотоксичность оценивалась с помощью МТТ-анализа после воздействия различных концентраций ДХА в течение 72 ч. (B) Изменение биоэнергетики под воздействием ДХА в клетках HNC. Измерения гликолиза и OXPHOS (%) были описаны в разделе «Материалы и методы». (C) Корреляция между гликолизом и IC50 цисплатина в группе клеточных линий HNC. IC50 рассчитывали с помощью МТТ-анализа в трех независимых экспериментах, каждый из которых проводился с использованием трех экземпляров образцов. Корреляция оценивалась с помощью линейной регрессии. (D) Корреляция между биоэнергетическими изменениями и апоптозом в клетках HNC. В анализе апоптоза измеряли аннексин V-положительные апоптотические фракции после воздействия 20 мМ DCA в течение 72 часов.
Экспрессия PDK2 связана с устойчивостью к цисплатину в HNC
Цитотоксическое действие цисплатина было проверено на культивируемых клеточных линиях HN4-cisR и HN9-cisR и родительских раковых клетках (рис. 2А). Устойчивые к цисплатину клетки HN4-cisR и HN9-cisR показали 12-кратное и 18-кратное увеличение IC50, соответственно, по сравнению с соответствующими родительскими линиями. Вестерн-блот анализ показал, что HK2 и PDK2 были высоко экспрессированы в клетках HN4-cisR и HN9-cisR по сравнению с соответствующими родительскими клетками, в то время как экспрессия PDHE1α была низкой в клетках, устойчивых к цисплатину (рис. 2B). Уровень экспрессии мРНК PDK2 также был выше в резистентных клетках, чем в чувствительных (P < 0,01) (рис. 2C). DCA подавлял рост цисплатин-резистентных клеток HNC в той же степени, что и цисплатин-чувствительных клеток HNC (рис. 2D). Вестерн-блот анализ показал, что DCA значительно снизил уровень PDK2 и фосфо-PDHE1α (pPDHE1α), но повысил уровень проапоптотических белков, включая расщепленный PARP (cPARP) и PUMA, а также p21 в HN4-cisR и HN9-cisR (рис. 2E). Это говорит о том, что DCA может эффективно подавлять рост цисплатин-резистентных клеток HNC, а также чувствительных к цисплатину клеток HNC.
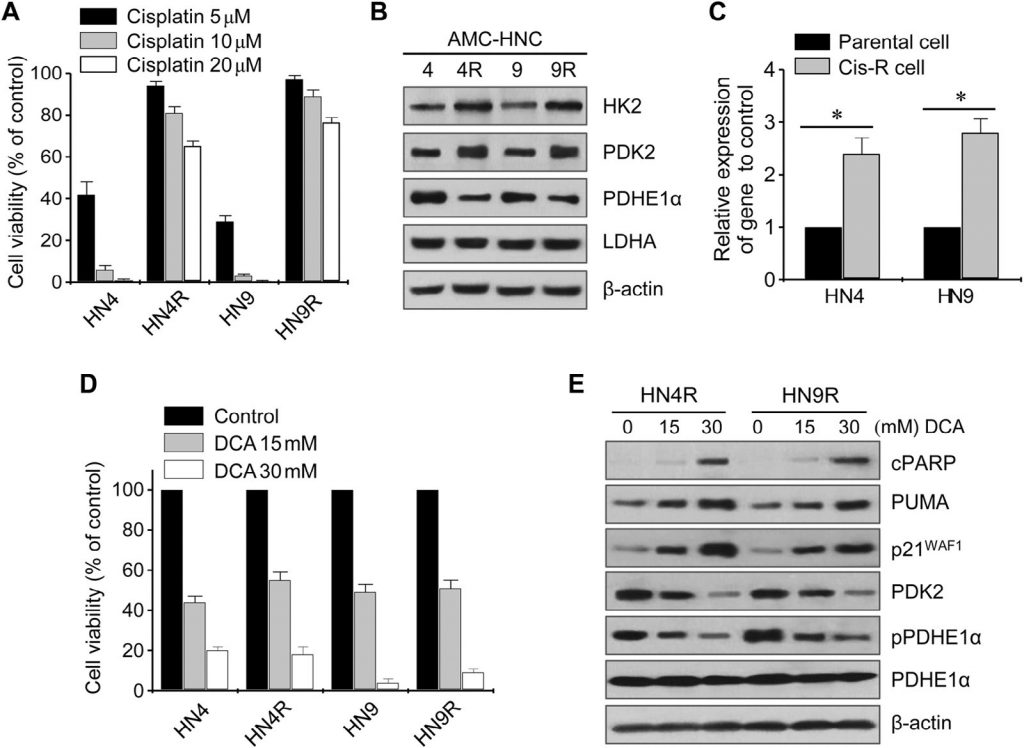 Рис. 2. Экспрессия PDK2 связана с устойчивостью к цисплатину в НЯК. (А) Жизнеспособность клеток оценивалась с помощью МТТ-анализа после воздействия цисплатина в течение 72 ч. (B) Вестерн-блот анализ показывает различные уровни белков гексокиназы 2 (HK2), киназы пируватдегидрогеназы 2 (PDK2), пируватдегидрогеназы (PDH) E1α (PDHE1α) и лактатдегидрогеназы-A (LDHA) в необработанных клетках HN4 (HN4R) и HN9 (HN9R), устойчивых к цисплатину, и в их родительских клетках. (C) Изменение уровня экспрессии гена PDK2 между клетками, устойчивыми к цисплатину (cis-R), и их родительскими клетками. * обозначает P <0,01 относительно родительских клеток HNC. (D) Жизнеспособность клеток оценивали с помощью МТТ-анализа после воздействия 15 и 30 мМ DCA в течение 72 ч. Столбики ошибок представляют собой среднюю квадратичную ошибку трех независимых экспериментов, каждый из которых проводился с использованием трех экземпляров образцов. (E) Вестерн-блот анализ выявляет изменения в уровнях расщепленного PARP, PUMA, p21, PDK2, phosho-PDHE1α (pPDHE1α) и PDHE1α в цисплатин-устойчивых клетках HN4R и HN9R, подвергнутых воздействию ДКА в течение 24 ч. Уровень β-актина оценивался в качестве контроля загрузки.
Рис. 2. Экспрессия PDK2 связана с устойчивостью к цисплатину в НЯК. (А) Жизнеспособность клеток оценивалась с помощью МТТ-анализа после воздействия цисплатина в течение 72 ч. (B) Вестерн-блот анализ показывает различные уровни белков гексокиназы 2 (HK2), киназы пируватдегидрогеназы 2 (PDK2), пируватдегидрогеназы (PDH) E1α (PDHE1α) и лактатдегидрогеназы-A (LDHA) в необработанных клетках HN4 (HN4R) и HN9 (HN9R), устойчивых к цисплатину, и в их родительских клетках. (C) Изменение уровня экспрессии гена PDK2 между клетками, устойчивыми к цисплатину (cis-R), и их родительскими клетками. * обозначает P <0,01 относительно родительских клеток HNC. (D) Жизнеспособность клеток оценивали с помощью МТТ-анализа после воздействия 15 и 30 мМ DCA в течение 72 ч. Столбики ошибок представляют собой среднюю квадратичную ошибку трех независимых экспериментов, каждый из которых проводился с использованием трех экземпляров образцов. (E) Вестерн-блот анализ выявляет изменения в уровнях расщепленного PARP, PUMA, p21, PDK2, phosho-PDHE1α (pPDHE1α) и PDHE1α в цисплатин-устойчивых клетках HN4R и HN9R, подвергнутых воздействию ДКА в течение 24 ч. Уровень β-актина оценивался в качестве контроля загрузки.
DCA индуцирует накопление ROS в клетках HNC
Изменение клеточной ROS под воздействием DCA оценивалось методом проточной цитометрии с использованием редокс-чувствительного флуоресцентного зонда DCF-DA. Воздействие DCA вызвало значительное повышение уровня ROS в клетках HNC (P < 0,01), которое блокировалось совместным воздействием NAC или ингибитором SOD диэтил-дитиокарбаматом (рис. 3A). Гликолиз значительно увеличился в цисплатин-устойчивых клетках HNC по сравнению с родительскими клетками (P < 0,01), но DCA вызвал значительное снижение гликолиза и увеличение окислительного фосфорилирования как в чувствительных к цисплатину, так и в цисплатин-устойчивых клетках HNC (P < 0,01) (рис. 3B). Мембранный потенциал митохондрий (ΔΨm) измеряли с помощью TMRM, чувствительного к напряжению митохондрий красителя на основе родамина, а митохондриальную ROS (mROS, супероксид митохондрий) измеряли с помощью MitoSOX red. В клетках HNC, обработанных DCA, наблюдалось снижение ΔΨm и увеличение mROS (P < 0,01) (рис. 3C). Изменение ΔΨm в цисплатин-устойчивых клетках HNC при воздействии DCA блокировалось предварительной обработкой NAC (рис. 3Д). Это говорит о том, что DCA может вызывать накопление ROS в клетках HNC путем активации окислительного фосфорилирования.
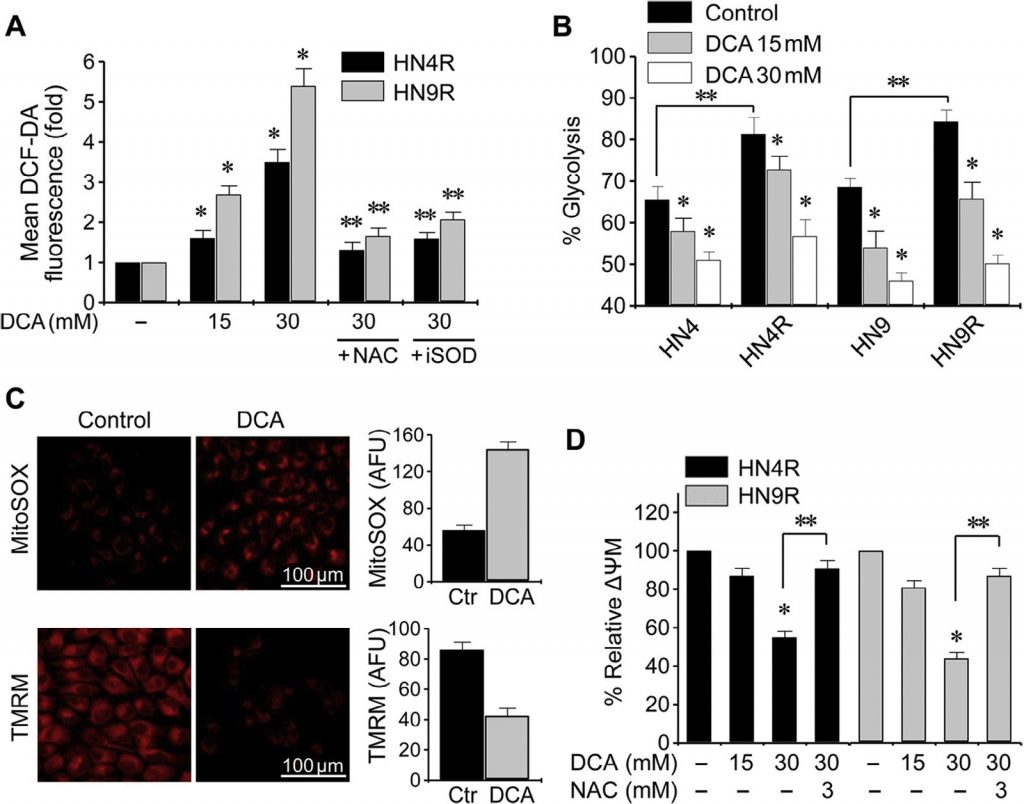 Рис. 3. DCA индуцирует внутриклеточное накопление ROS в клетках HNC. (A) Повышение уровня ROS под действием DCA. Устойчивые к цисплатину клетки HN4R и HN9R подвергали воздействию 15 или 30 мМ DCA в течение 24 ч. Клетки также предварительно обрабатывали ингибитором ROS N-ацетил-цистеином(NAC, 3 мМ) или ингибитором супероксиддисмутазы (iSOD) диэтил-дитиокарбаматом (10 мкМ). Уровни ROS измерялись методом проточной цитометрии с использованием DCF-DA и показаны как изменения в разах по сравнению с контрольным (базальным) уровнем. Все значения являются средними ± S.E. трех независимых экспериментов. * обозначает P <0,01 относительно контроля, и ** обозначает P <0,01 относительно 30 мМ DCA. (B) Изменение биоэнергетики под действием ДКА в клетках HNC. * обозначает P <0,05 по сравнению с контролем, и ** обозначает P <0,01 по сравнению с родительскими клетками, чувствительными к цисплатину. (C) Изменение реактивных форм кислорода (ROS, т.е. супероксид), измеренных с помощью mitoSOX, и мембранного потенциала митохондрий (ΔΨm, красное окрашивание) при воздействии 20 мМ DCA по сравнению с необработанным контролем (ctr). Произвольные единицы флуоресценции (AFU) в контрольных и обработанных ДКА клетках. (D) Изменения ΔΨm в клетках HN4R и HN9R после 36 ч воздействия ДКА. ΔΨm измеряли с помощью тетраметилродамин этилового эфира и анализировали методом проточной цитометрии. Средняя интенсивность флуоресценции в каждой группе лечения была нормализована по отношению к контрольной группе. Планки ошибок представляют собой среднюю квадратичную ошибку трех независимых экспериментов, каждый из которых проводился с использованием трех экземпляров образцов. * обозначает P <0,01 по сравнению с контролем. ** обозначает P <0,01 по сравнению с DCA или DCA плюс NAC.
Рис. 3. DCA индуцирует внутриклеточное накопление ROS в клетках HNC. (A) Повышение уровня ROS под действием DCA. Устойчивые к цисплатину клетки HN4R и HN9R подвергали воздействию 15 или 30 мМ DCA в течение 24 ч. Клетки также предварительно обрабатывали ингибитором ROS N-ацетил-цистеином(NAC, 3 мМ) или ингибитором супероксиддисмутазы (iSOD) диэтил-дитиокарбаматом (10 мкМ). Уровни ROS измерялись методом проточной цитометрии с использованием DCF-DA и показаны как изменения в разах по сравнению с контрольным (базальным) уровнем. Все значения являются средними ± S.E. трех независимых экспериментов. * обозначает P <0,01 относительно контроля, и ** обозначает P <0,01 относительно 30 мМ DCA. (B) Изменение биоэнергетики под действием ДКА в клетках HNC. * обозначает P <0,05 по сравнению с контролем, и ** обозначает P <0,01 по сравнению с родительскими клетками, чувствительными к цисплатину. (C) Изменение реактивных форм кислорода (ROS, т.е. супероксид), измеренных с помощью mitoSOX, и мембранного потенциала митохондрий (ΔΨm, красное окрашивание) при воздействии 20 мМ DCA по сравнению с необработанным контролем (ctr). Произвольные единицы флуоресценции (AFU) в контрольных и обработанных ДКА клетках. (D) Изменения ΔΨm в клетках HN4R и HN9R после 36 ч воздействия ДКА. ΔΨm измеряли с помощью тетраметилродамин этилового эфира и анализировали методом проточной цитометрии. Средняя интенсивность флуоресценции в каждой группе лечения была нормализована по отношению к контрольной группе. Планки ошибок представляют собой среднюю квадратичную ошибку трех независимых экспериментов, каждый из которых проводился с использованием трех экземпляров образцов. * обозначает P <0,01 по сравнению с контролем. ** обозначает P <0,01 по сравнению с DCA или DCA плюс NAC.
ДКА способствует остановке клеточного цикла и апоптозу в клетках HNC
Клоногенный анализ привел к заметному снижению числа колоний HN4-cisR и HN9-cisR при воздействии ДКА (P < 0,01) (рис. 4A ). Проточная цитометрия с окрашиванием йодистым пропидием показала значительное изменение клеточного цикла в клетках HNC: вогонин увеличил популяцию апоптотических клеток sub-G1 (P < 0,05), и этот эффект блокировался при совместном воздействии с NAC (рис. 4B ). Вестерн-блот анализ показал, что DCA снижал pPDHE1α, но увеличивал cPARP, p21, фосфо-p53 и расщепленную каспазу-3 (cCasp3) в зависимости от времени (рис. 4C ). Активация PDHE1α путем нокдауна PDK2 в клетках AMC-HN4-cisR не привела к значительному увеличению проапоптотических белков cPARP и cCasp3 (рис. 4D ); однако нокдаун PDHE1α снизил экспрессию p21 и фосфорилирование p53. Проточная цитометрия с использованием йодистого пропидия и окрашивания аннексином-V показала эффективную индукцию апоптоза и гибели клеток в цисплатин-устойчивых клетках HNC под действием DCA; эффект снижался при совместном воздействии DCA и антиоксиданта NAC или пан-каспазного ингибитора Z-VAD-fmk (рис. 4E). Это было подтверждено измерением активности каспазы, которая значительно увеличивалась под действием ДКА в зависимости от концентрации (рис. 4F).
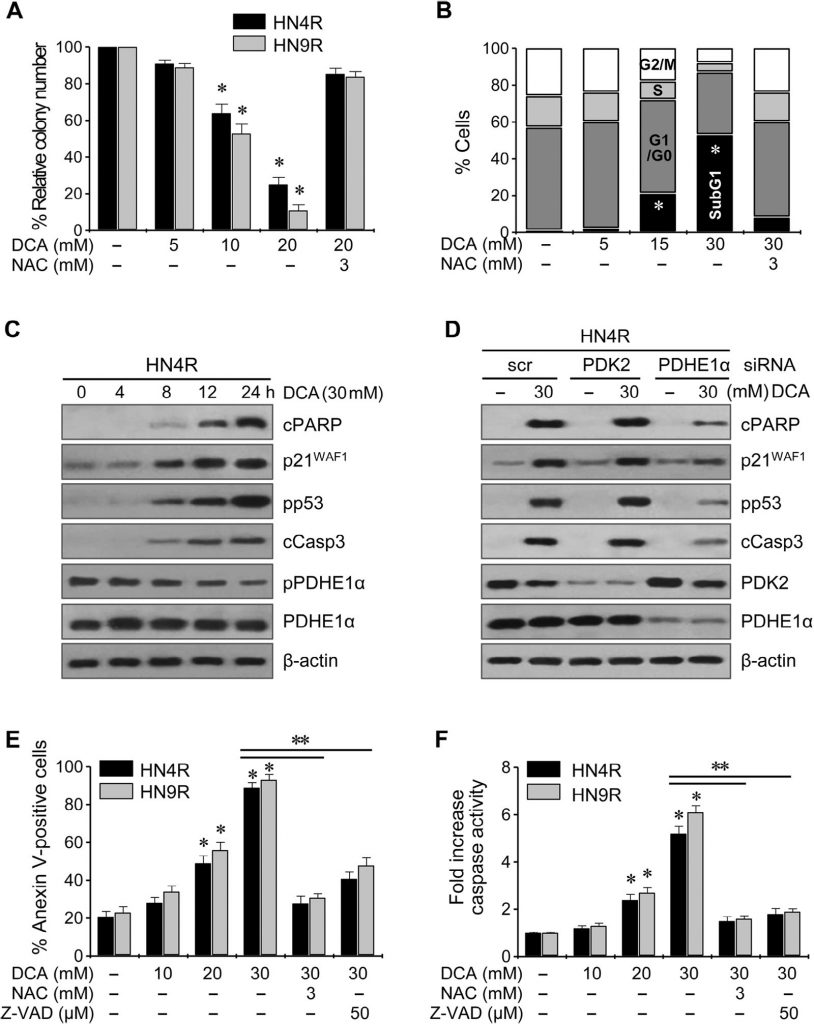 Рис. 4. DCA индуцирует гибель клеток и изменения клеточного цикла в клетках HNC. (A) Клоногенный анализ устойчивых к цисплатину раковых клеточных линий, подвергнутых воздействию DCA. Раковые клетки AMC-HN4R и HN9R подвергались воздействию DCA в течение 72 ч. Столбики ошибок представляют собой средние квадратичные значения трех независимых экспериментов, каждый из которых проводился в трех экземплярах. * обозначает P <0,01. (B) Анализ клеточного цикла при воздействии ДКА. Клетки AMC-HN4R, подвергнутые воздействию DCA в течение 72 ч, окрашивали йодистым пропидием и подвергали проточному цитометрическому анализу. * обозначает P <0,05. (C) Вестерн-блот анализ выявил изменения в уровнях расщепленного PARP (cPARP), p21WAF1, фосфо-p53-ser 15 (pp53), расщепленной каспазы 3 (cCasp3), pPDHE1α и PDHE1α. Клеточные экстракты были получены после воздействия на клетки AMC-HN4R 30 мМ DCA. (D) Влияние DCA и генетического ингибирования PDK2 и PDHE1α на проапоптотические белки, расщепленный PARP и расщепленную каспазу 3, а также на p21WAF1. Белки были измерены с помощью Вестерн-блот анализа в клетках AMC-HN4R, подвергнутых воздействию 30 мМ DCA. Раковые клетки были трансфицированы скремблированной siRNA (scr), PDK2 siRNA или PDHE1α siRNA в течение 48 ч до воздействия 30 мМ DCA. (E) Анализ апоптоза в клетках AMC-HN4R и HN9R, подвергнутых воздействию ДКА. Клетки подвергали воздействию ДКА в течение 72 ч, после чего измеряли апоптотические фракции, позитивные по аннексину V. (F) Повышение активности каспазы при обработке ДКА. Столбики ошибок представляют собой среднюю квадратичную ошибку трех повторов. * и ** обозначают P <0,01 относительно контроля и 30 мМ DCA, соответственно. Перед воздействием 30 мМ ДКА клетки также предварительно обрабатывали 3 мМ NAC или 50 мкМ пан-каспазного ингибитора Z-VAD-fmk.
Рис. 4. DCA индуцирует гибель клеток и изменения клеточного цикла в клетках HNC. (A) Клоногенный анализ устойчивых к цисплатину раковых клеточных линий, подвергнутых воздействию DCA. Раковые клетки AMC-HN4R и HN9R подвергались воздействию DCA в течение 72 ч. Столбики ошибок представляют собой средние квадратичные значения трех независимых экспериментов, каждый из которых проводился в трех экземплярах. * обозначает P <0,01. (B) Анализ клеточного цикла при воздействии ДКА. Клетки AMC-HN4R, подвергнутые воздействию DCA в течение 72 ч, окрашивали йодистым пропидием и подвергали проточному цитометрическому анализу. * обозначает P <0,05. (C) Вестерн-блот анализ выявил изменения в уровнях расщепленного PARP (cPARP), p21WAF1, фосфо-p53-ser 15 (pp53), расщепленной каспазы 3 (cCasp3), pPDHE1α и PDHE1α. Клеточные экстракты были получены после воздействия на клетки AMC-HN4R 30 мМ DCA. (D) Влияние DCA и генетического ингибирования PDK2 и PDHE1α на проапоптотические белки, расщепленный PARP и расщепленную каспазу 3, а также на p21WAF1. Белки были измерены с помощью Вестерн-блот анализа в клетках AMC-HN4R, подвергнутых воздействию 30 мМ DCA. Раковые клетки были трансфицированы скремблированной siRNA (scr), PDK2 siRNA или PDHE1α siRNA в течение 48 ч до воздействия 30 мМ DCA. (E) Анализ апоптоза в клетках AMC-HN4R и HN9R, подвергнутых воздействию ДКА. Клетки подвергали воздействию ДКА в течение 72 ч, после чего измеряли апоптотические фракции, позитивные по аннексину V. (F) Повышение активности каспазы при обработке ДКА. Столбики ошибок представляют собой среднюю квадратичную ошибку трех повторов. * и ** обозначают P <0,01 относительно контроля и 30 мМ DCA, соответственно. Перед воздействием 30 мМ ДКА клетки также предварительно обрабатывали 3 мМ NAC или 50 мкМ пан-каспазного ингибитора Z-VAD-fmk.
DCA сенсибилизирует цисплатин-устойчивые клетки HNC к цисплатину in vitro и in vivo
Цисплатин (10 мкМ) не вызывал значительной цитотоксичности или экспрессии апоптотических белков в цисплатин-устойчивых линиях HNC HN4-cisR и HN9-cisR по сравнению с родительскими чувствительными к цисплатину линиями HN4 и HN9 (рис.
5А).
5A); однако DCA вызывал заметное снижение выживаемости в цисплатин-устойчивых клетках HNC (P < 0,05), которое блокировалось предварительной обработкой NAC. DCA индуцировал экспрессию апоптотических белков и увеличивал цисплатин-индуцированную цитотоксичность и экспрессию апоптотических белков в клетках HN4-cisR (рис.
B
). В комбинации DCA увеличивал цитотоксичность цисплатина в клетках HN4-cisR за счет повышения активности каспаз до степени, превышающей сумму эффектов каждого из агентов в отдельности (CI < 1, P < 0,01), которые ослаблялись при глушении гена PDHE1α (рис. 5C). Уровень ΔΨm был выше в клетках HN4-cisR, устойчивых к цисплатину, чем в чувствительных к цисплатину родительских клетках HN4 (ΔΨm, средняя интенсивность флуоресценции [MFI]: 1 ± 0 против 0,54 ± 0,09, P < 0,001) и снижался под действием DCA или комбинации цисплатина и DCA, что ослаблялось глушением гена PDHE1α (рис. 5D).
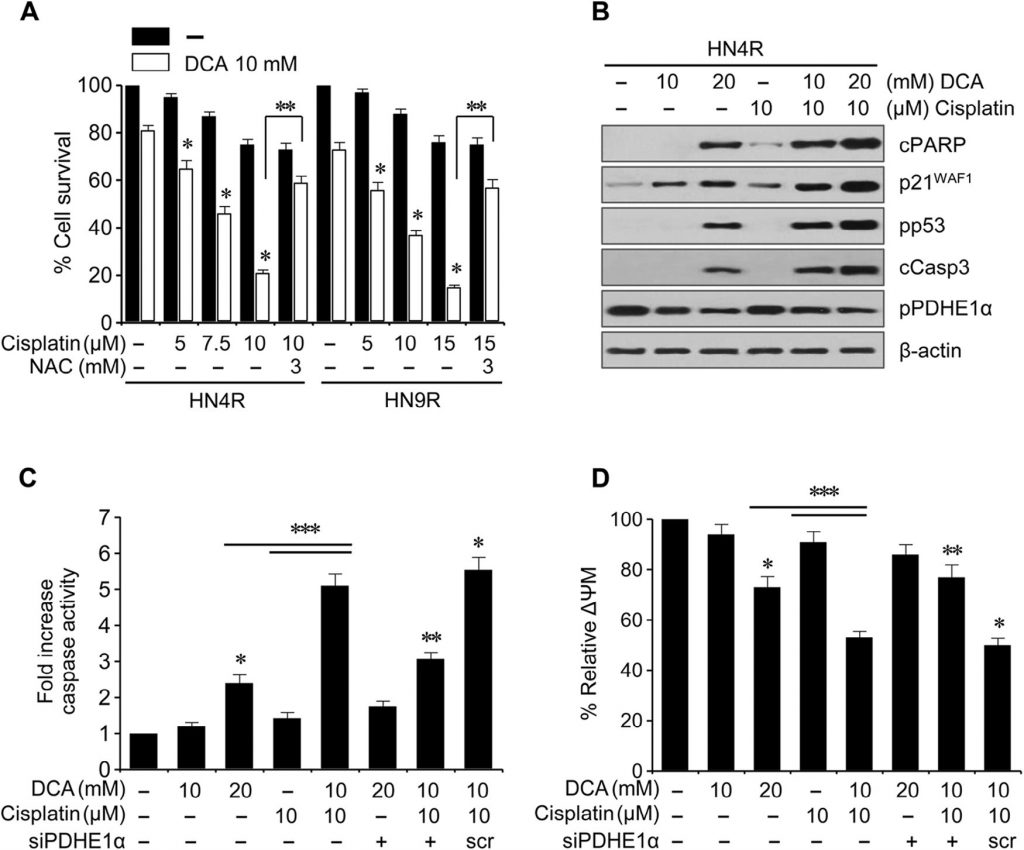 Рис. 5. DCA сенсибилизировал цисплатин-резистентные клетки HNC к цисплатину. (А) Жизнеспособность клеток оценивали по исключению трипанового синего после воздействия цисплатина (CDDP), DCA или их комбинации. Цитотоксический эффект DCA блокировался антиоксидантом N-ацетил-цистеином(NAC, 3 мМ). Планки ошибок представляют собой среднюю квадратичную ошибку трех независимых экспериментов, каждый из которых проводился с использованием трех экземпляров образцов. * обозначает P <0,01 по сравнению с контролем, и ** обозначает P <0,01 по сравнению с предварительной обработкой NAC. (B) Вестерн-блоттинг, показывающий увеличение чувствительности к цисплатину (CDDP) под действием DCA. Устойчивые к цисплатину клетки AMC-HN4R обрабатывали DCA, цисплатином или обоими препаратами в течение 72 ч. (C) Повышение уровня каспаз после 72-часового воздействия DCA, цисплатина и PDHE1α siRNA. Клетки HN4R подвергались воздействию DCA, цисплатина или комбинации обоих препаратов, после чего измерялась активность каспазы.(D) Изменения ΔΨm в клетках HN4R после 36-часового воздействия DCA, цисплатина или комбинации обоих препаратов. ΔΨm измеряли с помощью этилового эфира тетраметилродамина и анализировали методом проточной цитометрии. Медиана интенсивности флуоресценции в каждой группе лечения была нормализована по отношению к контрольной группе. Планки ошибок представляют собой среднюю квадратичную ошибку трех независимых экспериментов, каждый из которых проводился с использованием трех экземпляров образцов. * и ** означают P <0,01 по сравнению с контролем и скремблированной (scr) siRNA, соответственно. *** обозначает P <0,01 по сравнению с DCA или цисплатином.
Рис. 5. DCA сенсибилизировал цисплатин-резистентные клетки HNC к цисплатину. (А) Жизнеспособность клеток оценивали по исключению трипанового синего после воздействия цисплатина (CDDP), DCA или их комбинации. Цитотоксический эффект DCA блокировался антиоксидантом N-ацетил-цистеином(NAC, 3 мМ). Планки ошибок представляют собой среднюю квадратичную ошибку трех независимых экспериментов, каждый из которых проводился с использованием трех экземпляров образцов. * обозначает P <0,01 по сравнению с контролем, и ** обозначает P <0,01 по сравнению с предварительной обработкой NAC. (B) Вестерн-блоттинг, показывающий увеличение чувствительности к цисплатину (CDDP) под действием DCA. Устойчивые к цисплатину клетки AMC-HN4R обрабатывали DCA, цисплатином или обоими препаратами в течение 72 ч. (C) Повышение уровня каспаз после 72-часового воздействия DCA, цисплатина и PDHE1α siRNA. Клетки HN4R подвергались воздействию DCA, цисплатина или комбинации обоих препаратов, после чего измерялась активность каспазы.(D) Изменения ΔΨm в клетках HN4R после 36-часового воздействия DCA, цисплатина или комбинации обоих препаратов. ΔΨm измеряли с помощью этилового эфира тетраметилродамина и анализировали методом проточной цитометрии. Медиана интенсивности флуоресценции в каждой группе лечения была нормализована по отношению к контрольной группе. Планки ошибок представляют собой среднюю квадратичную ошибку трех независимых экспериментов, каждый из которых проводился с использованием трех экземпляров образцов. * и ** означают P <0,01 по сравнению с контролем и скремблированной (scr) siRNA, соответственно. *** обозначает P <0,01 по сравнению с DCA или цисплатином.
Эти результаты были далее изучены на мышиных моделях опухолевых ксенотрансплантатов in vivo. Атимичные мыши BALB/c, несущие опухоли AMC-HN4-cisR или HN9-cisR, получали DCA, цисплатин, DCA плюс цисплатин или транспортное средство. Комбинация цисплатина и DCA синергетически подавляла рост опухоли (рис. 6A). Визуализация роста опухоли in vivo проводилась с помощью ПЭТ с 18 F-ФДГ на 21 день лечения. Очаговые поглощения 18 F-ФДГ наблюдались в местах имплантации опухоли, где измерялось максимальное стандартизированное поглощение (SUVmax) и метаболический объем опухоли при SUV 2.0 (MTV2.0). SUVmax не отличался между группами лечения (P > 0,1), но MTV2.0 был значительно ниже в группах, получавших DCA и комбинацию, чем в других группах (P < 0,05) (рис. 6B). Анализ апоптоза in situ показал, что TUNEL-положительные апоптотические тельца чаще встречались в опухолях, получавших DCA и цисплатин плюс DCA, чем в опухолях, получавших транспортное средство (P < 0,01) (рис. 6C). Вестерн-блот анализ опухолевых тканей показал, что уровень апоптотических белков увеличился в большей степени в клетках HN4-cisR, обработанных комбинацией цисплатина и DCA, чем в опухолях, обработанных одним агентом (рис. 6D).
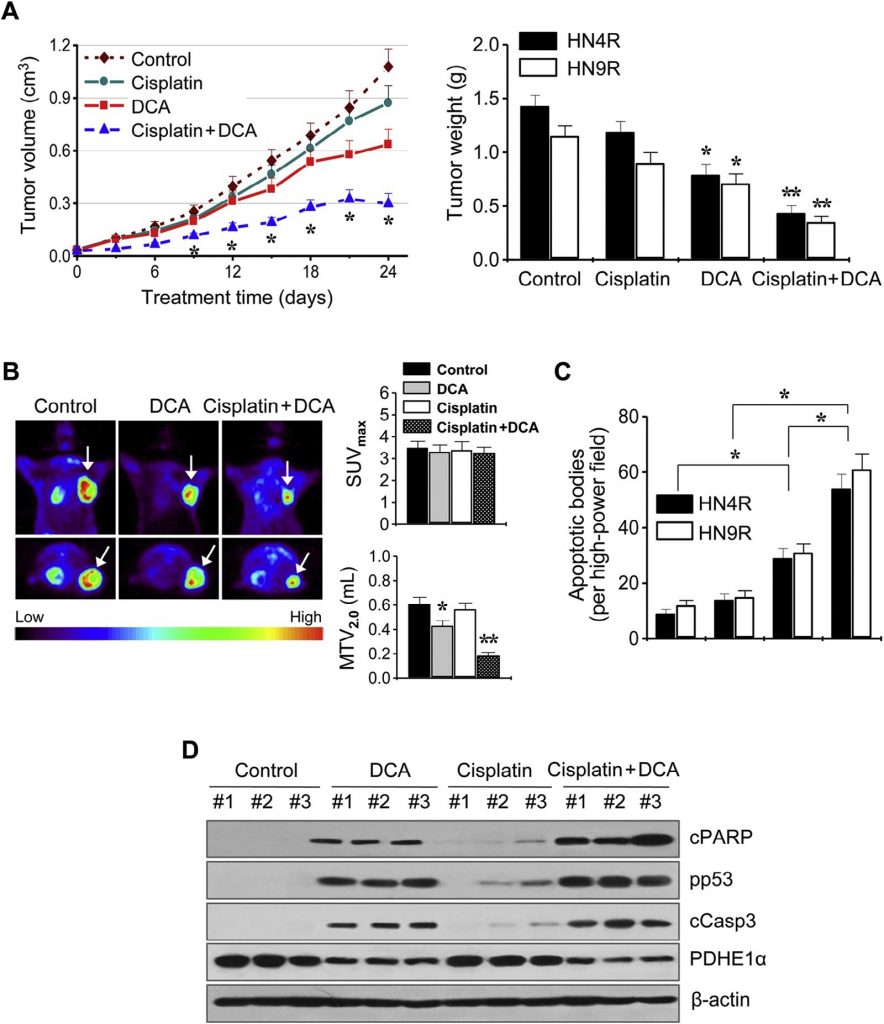 Рис. 6. DCA сенсибилизирует цисплатин-резистентные клетки HNC к цисплатину in vivo. (A) Противоопухолевый эффект DCA и цисплатина в модели опухолевого ксенотрансплантата на мышах. Голым мышам вводили 5 ×106 клеток AMC-HN4R и HN9R во фланг. Лечение транспортным средством, цисплатином (CDDP), DCA или комбинацией цисплатина и вогонина начиналось после того, как имплантированные опухолевые клетки образовывали пальпируемые узелки. Каждая группа включала десять мышей. Столбики ошибок представляют S.E. * и ** обозначают P <0,05 и P <0,01 по сравнению с контролем (ctr), соответственно. (B) Визуализация опухоли in vivo с помощью позитронно-эмиссионной томографии (ПЭТ) с 18F-фтордезоксиглюкозой. ПЭТ-сканирование проводилось через 3 недели после начала лечения. Максимальный стандартизированный объем поглощения (SUVmax) и метаболический объем опухоли (MTV2.0) были получены в опухолях (стрелки), и средние значения сравнивались между различными группами лечения. * и ** обозначают P <0,05 и P <0,01 по сравнению с контролем, соответственно. (C) Количественная оценка по результатам анализа TUNEL in situ в срезах опухолей из каждой группы. TUNEL-позитивные апоптотические тельца подсчитывали вслепую в десяти случайно выбранных мощных полях. Столбики ошибок представляют S.E. Двухсторонний t-тест Стьюдента, * обозначает P <0,01. (D) Вестерн-блот анализ расщепленного PARP, фосфо-p53-ser15, расщепленной каспазы 3 и белков PDHE1α, полученных из опухолей, обработанных транспортным средством, DCA, цисплатином или комбинацией обоих препаратов. β-актин служил в качестве внутреннего контроля нагрузки.
Рис. 6. DCA сенсибилизирует цисплатин-резистентные клетки HNC к цисплатину in vivo. (A) Противоопухолевый эффект DCA и цисплатина в модели опухолевого ксенотрансплантата на мышах. Голым мышам вводили 5 ×106 клеток AMC-HN4R и HN9R во фланг. Лечение транспортным средством, цисплатином (CDDP), DCA или комбинацией цисплатина и вогонина начиналось после того, как имплантированные опухолевые клетки образовывали пальпируемые узелки. Каждая группа включала десять мышей. Столбики ошибок представляют S.E. * и ** обозначают P <0,05 и P <0,01 по сравнению с контролем (ctr), соответственно. (B) Визуализация опухоли in vivo с помощью позитронно-эмиссионной томографии (ПЭТ) с 18F-фтордезоксиглюкозой. ПЭТ-сканирование проводилось через 3 недели после начала лечения. Максимальный стандартизированный объем поглощения (SUVmax) и метаболический объем опухоли (MTV2.0) были получены в опухолях (стрелки), и средние значения сравнивались между различными группами лечения. * и ** обозначают P <0,05 и P <0,01 по сравнению с контролем, соответственно. (C) Количественная оценка по результатам анализа TUNEL in situ в срезах опухолей из каждой группы. TUNEL-позитивные апоптотические тельца подсчитывали вслепую в десяти случайно выбранных мощных полях. Столбики ошибок представляют S.E. Двухсторонний t-тест Стьюдента, * обозначает P <0,01. (D) Вестерн-блот анализ расщепленного PARP, фосфо-p53-ser15, расщепленной каспазы 3 и белков PDHE1α, полученных из опухолей, обработанных транспортным средством, DCA, цисплатином или комбинацией обоих препаратов. β-актин служил в качестве внутреннего контроля нагрузки.
Обсуждение
Повышенный гликолиз, который часто встречается при раке, тесно связан с нарушением митохондрий или дефектным окислительным фосфорилированием и способствует терапевтической резистентности [18,19]. Аэробный гликолиз был связан с устойчивостью к химиотерапии [20] или радиотерапии [21]. В настоящем исследовании клеточные линии, устойчивые к цисплатину, показали повышенный гликолиз, что указывает на биохимическую связь между гликолизом и химиорезистентностью. Устойчивые к лекарствам клетки испытывают большую потребность в АТФ, чем нормальные клетки, для поддержания клеточного гомеостаза и активации путей выживания, которые позволяют избежать гибели клеток при генотоксическом стрессе [20]. Устойчивые раковые клетки увеличивают гликолиз для быстрой выработки АТФ, чтобы удовлетворить внутриклеточные потребности. Это было четко обнаружено в наших клетках, устойчивых к цисплатину, по сравнению с родительскими чувствительными клетками, что указывает на то, что лекарственная устойчивость в HNC напрямую связана с увеличением гликолиза. Это означает, что изменение биоэнергетики раковых клеток в сторону увеличения гликолиза связано с химиорезистентностью [14,22]. Таким образом, уровень гликолиза в раковых клетках человека может быть биомаркером химиорезистентности и требует клинического подтверждения в раковых тканях человека.
В настоящем исследовании показано, что DCA смещает генерацию энергии с гликолиза на митохондриальное окисление глюкозы в HNC. Это вызванное DCA изменение метаболического сдвига в сторону гликолиза устраняет пролиферативное преимущество раковых клеток и в конечном итоге приводит к их гибели [23]. DCA, являясь структурным аналогом пирувата, ингибирует PDK и реактивирует PDH, фермент, входящий в комплекс, который преобразует пируват в ацетил-КоА, основной субстрат цикла Кребса [10,23]. Поскольку большинство типов рака создают гипоксическую среду, раковые клетки полагаются на анаэробный гликолиз в качестве основного источника энергии. Активация гипоксия-индуцибельного фактора (HIF) индуцирует митохондриальную PDK [24]. Митохондриальная активность PDH в раке блокируется PDK, что приводит к снижению доступности ацетил-КоА для митохондриального окисления глюкозы [24]. Повышенная экспрессия PDK связана с лекарственной устойчивостью при раке [25,26]. Регуляция PDK2 в результате митохондриальных мутаций и стабилизации HIF1α также наблюдается в клетках HNC [27]. В настоящем исследовании также была подтверждена связь между сверхэкспрессией PDK2 и устойчивостью к цисплатину в клетках HNC. Эти данные указывают на важность PDK как новой молекулярной мишени в терапии рака [10]. Предыдущие данные показали, что генетическое или фармакологическое ингибирование PDK изменяет биоэнергетику рака и восстанавливает митохондрий-зависимый апоптоз в раковых клетках [11,13]. Поэтому ДКК эффективен для преодоления резистентности к лечению в раковых опухолях с агрессивным фенотипом in vitro и in vivo[12-14].
Активация PDH под действием DCA вызывает накопление mROS в раковых клетках. В раковых клетках снижен митохондриальный метаболизм глюкозы, что приводит к снижению активности электронно-транспортной цепи (ЭТЦ) и уменьшению mROS [10], [28]. Митохондриальное ремоделирование ΔΨm гиперполяризации и снижение выработки mROS в раковых клетках приводит к устойчивости к апоптозу при генотоксическом стрессе. В устойчивых раковых клетках с более гликолитическим фенотипом HK2 приводит к транслокации на внешнюю митохондриальную мембрану и увеличению ΔΨm [29]. Ингибирование гликолиза и транслокации HK2 снижает ΔΨm рака и отменяет устойчивость к апоптозу [29]. В настоящем исследовании повышенная экспрессия HK2 и PDK2 и мембранный потенциал митохондрий также были обнаружены в устойчивых к цисплатину раковых клетках. DCA заставляет пируват поступать в митохондрии через активацию PDH, регулятора митохондриального окисления глюкозы, снижает ΔΨm и увеличивает mROS [13]. В настоящем исследовании DCA просто отменил повышенное митохондриальное ремоделирование в клетках HNC, устойчивых к цисплатину, что способствовало гибели раковых клеток. Активация митохондриального окисления глюкозы под действием DCA вызвала mROS и активацию митохондриальной сигнализации, что привело к активации p53 и связанных с ним проапоптотических путей и к гибели устойчивых к химиотерапии раковых клеток. В нашем исследовании фармакологическое ингибирование выработки mROS или каспаза-опосредованного апоптоза ослабляло цитотоксический эффект DCA в клетках HNC, что подтверждает известные механизмы действия препарата.
Доклинические и клинические исследования поддерживают применение ДКА у онкологических больных и у пациентов с молочнокислым ацидозом, связанным с митохондриальными заболеваниями [10,23]. В ряде исследований продемонстрировано цитотоксическое действие DCA, отдельно или в комбинации с другими препаратами, на различные опухоли, полученные из всех трех зародышевых слоев [23]. Предыдущее исследование показало, что глиобластома, один из самых агрессивных видов рака человека, сверхэкспрессирует PDK2 в раковых тканях, взятых у 49 пациентов, и регрессирует у пяти пациентов после лечения ДКА, что доказывает клиническую пользу ДКА в резистентных раковых опухолях [13]. В недавнем исследовании, посвященном HNC, сравнивался эффект DCA на трех клеточных линиях плоскоклеточной карциномы полости рта (OSCC) [15]. Клетки OSCC с дефицитом митохондриального окислительного фосфорилирования (т.е. высоким гликолизом) были более чувствительны к DCA, чем другие [15,30]. Настоящее исследование подтвердило, что клетки HNC с высокими биоэнергетическими изменениями были более чувствительны к DCA. Фактически, поскольку химиорезистентные раковые клетки имеют тенденцию к высокому гликолизу, эти клетки могут быть мишенью для ингибиторов метаболизма [31]. Таким образом, DCA является хорошим кандидатом для лечения раковых клеток с агрессивным фенотипом, включая химиорезистентные клетки HNC. Однако, поскольку потенциальные противораковые эффекты DCA все еще спорны, особенно в опухолях в условиях гипоксии, необходимы дальнейшие доклинические и клинические исследования DCA и рака [32]. Недавний систематический обзор показал, что DCA синергирует со многими стандартными противораковыми препаратами, а клинические испытания на ранней стадии свидетельствуют о том, что хронический DCA безопасен и хорошо переносится при пероральном приеме 12,5 мг/кг дважды в день [23].
заключение, наши данные свидетельствуют о том, что высокий уровень гликолиза и сверхэкспрессия PDK тесно связаны с устойчивостью к цисплатину в клетках HNC. DCA переключает биоэнергетику клеток НЯК на митохондриальное окислительное фосфорилирование. Это приводит к снижению ΔΨm и увеличению mROS, тем самым сенсибилизируя химиорезистентные клетки НЯК к цисплатину in vitro и in vivo. Эти данные дают основания для дальнейших доклинических и клинических исследований DCA, перспективного противоракового препарата, при НЯК с агрессивным фенотипом. Однако наше заключение следует принимать во внимание с осторожностью при противоположном сценарии. Дефектное митохондриальное окислительное фосфорилирование было связано с чувствительностью раковых клеток к состоянию низкой глюкозы, что может быть использовано в качестве биомаркера для терапии рака [37]. Раковая клетка также приспособлена для обеспечения пролиферации путем поглощения и включения питательных веществ в биомассу, а не для эффективного производства АТФ [38]. Поэтому доклинические и клинические эффекты DCA на HNC и другие виды рака должны быть изучены далее.