ВЛИЯНИЕ ДИХЛОРАЦЕТАТА НА РОСТ И МЕТАСТАЗИРОВАНИЕ КАРЦИНОМЫ ЛЕГКИХ ЛЬЮИСА
ВЛИЯНИЕ ДИХЛОРАЦЕТАТА НАТРИЯ НА РОСТ И МЕТАСТАЗИРОВАНИЕ КАРЦИНОМЫ ЛЕГКИХ ЛЬЮИСА
Д.Л. Колесник1, О.Н. Пясковская1, И.В. Бойчук1, О.И. Дасюкевич1, О.Р. Мельников1, А.С. Тарасов1, Г.И. Соляник1
1Институтэкспериментальной патологии, онкологии и радиобиологии имени
Р.Е. Кавецкого НАН Украины, Киев 03022, Украина.
Корреспонденция: Д.Л. Колесник, E-mail: deniskol@mail.ru
Поступила в редакцию: 20 марта 2015 г
Аннотация
Отличительной чертой злокачественных опухолей является чрезмерный гликолиз опухоли, даже в присутствии кислорода, что вызывает лактацидоз в микроокружении опухоли и способствует пролиферации и выживанию опухолевых клеток. По этой причине антиметаболические агенты, направленные на метаболизм опухолевых клеток, активно исследуются как перспективные противораковые препараты. Цель: изучить влияние лактацидоза на выживание клеток карциномы легких Льюиса (LLC) в условиях дефицита питательных субстратов in vitro и оценить противоопухолевую и антиметастатическую активность против LLC/R9 in vivo. Материалы и методы: В качестве экспериментальной опухолевой модели использовали вариант LLC/R9. Жизнеспособность опухолевых клеток определяли с помощью окрашивания трипановым синим. Уровень апоптоза подсчитывали с помощью красителя Hoechst 33258. Содержание лактата в опухолевой ткани оценивали ферментным методом с использованием лактатдегидрогеназы. Реактивные виды кислорода определяли с помощью 2,7-дихлорфлуоресцеин диацетата. Влияние дихлорацетата (ДХА) на рост и метастазирование LLC/R9 анализировали с помощью рутинных процедур. Оценка влияния ДХА на компоненты электронно-транспортной цепи (ЭТЦ) проводилась с помощью ЭПР. Результаты: Показано, что в условиях лактацидоза и дефицита глюкозы жизнеспособность клеток LLC/R9 in vitro была выше на 30% (р < 0,05), а уровень апоптоза был в три раза ниже (р < 0,05), чем эти показатели в условиях только дефицита глюкозы. У мышей с пересаженными опухолями LLC/R9, которых в течение 3 недель per os лечили DCA в общей дозе 1,5 г/кг массы тела, начиная со следующего дня после пересадки опухоли, объем первичной опухоли был всего на 30% меньше, чем в контрольной группе. В то же время количество и объем метастазов в легких у животных, получавших DCA, были на 59% (р < 0,05) и 94% (р < 0,05) ниже, соответственно, чем эти показатели в контрольной группе. Лечение DCA привело к увеличению содержания лактата в опухолевой ткани почти на 30% (р < 0,05) по сравнению с контролем, но не оказало существенного влияния на уровни комплексов гемового железа с NO (при gmed = 2,007) в белках митохондриального ЭТЦ и белков Fe-S кластера (при g = 1,94) в опухолевых клетках. Выводы: Показано, что лактацидоз значительно способствовал выживанию клеток LLC/R9 в условиях дефицита глюкозы in vitro. При развитии LLC/R9 in vivo DCA, как соединение с антилактацидозной активностью, не подавлял существенно рост первичной опухоли, но обладал значительной антиметастатической активностью.
Ключевые слова: дихлорацетат; гипоксическая радиочувствительность; рак молочной железы; реактивные виды кислорода
Сокращения: DCA — дихлорацетат, ETC — электронно-транспортная цепь; LLC/R9 — вариант карциномы легких Льюиса; PDH — пируватдегидрогеназа; PDK — киназа пируватдегидрогеназы.
ВВЕДЕНИЕ
Хорошо известно, что лактацидоз, большое накопление лактата и снижение рН, является основной характеристикой метаболического микроокружения опухолевых клеток in vitro и in vivo. Ранее лактацидоз рассматривался как балластный продукт метаболизма опухолевых клеток. Однако недавно было показано, что он может использоваться опухолевыми клетками в качестве эффективного энергетического топлива и быть одним из факторов, ответственных за устойчивость опухоли к дефициту глюкозы [1-3]. Как мы показали на примере клеток карциномы легких Льюиса (LLC)/R9, варианта LLC, чувствительного к антиангиогенной терапии рака [4,5], лактацидоз может способствовать выживанию опухолевых клеток в условиях дефицита питания. Такие условия были созданы путем длительной инкубации опухолевых клеток без замены культуральной среды (модель «непитательной культуры») [6]. Изучение кинетики роста опухолевых клеток в условиях «непитательной культуры» показало, что на фоне полного отсутствия глюкозы в инкубационной среде на 7-8 день роста клеток количество жизнеспособных клеток не падало ниже третьей части от максимума, зарегистрированного на 3-4 день, и оставалось практически на этом уровне до10 дня. Высокая выживаемость клеток LLC/R9 в условиях «некормленой культуры» была связана, в частности, со способностью этих клеток к макроавтофагии. Однако нельзя исключить, что способность клеток LLC/R9 к адаптации к дефициту питательных субстратов определялась лактацидозом, развившимся вследствие длительного культивирования опухолевых клеток без замены инкубационной среды.
Если лактацидоз способен повышать выживаемость опухолевых клеток, то соединения, подавляющие образование лактацидоза в опухолевом микроокружении, в частности, дихлорацетат (ДХА) как соединение с антилактацидозной активностью, должны проявлять противоопухолевую активность. Настоящее исследование было направлено на проверку этого предположения.
Известно, что DCA является ингибитором киназы пируватдегидрогеназы (PDK), поэтому его рассматривают как негативный регулятор ферментов митохондриального комплекса пируватдегидрогеназы (PDH), который играет ключевую роль в регуляции трикарбоновых кислот и окислительного фосфорилирования [7]. Если комплекс PDH фосфорилирован, вход пирувата в цикл Кребса ингибируется, поэтому активируется гликолиз. Благодаря ингибированию PDK, DCA способен привести к непрямой активации ферментов комплекса PDH и, соответственно, вызвать сдвиг энергетического баланса клетки от гликолиза в сторону активации окислительного фосфорилирования. Поэтому DCA широко используется для коррекции лактации, вызванной высокой интенсивностью гликолиза или дефектом клеточного дыхания.
Согласно литературным данным, способность ДКА активировать окислительное фосфорилирование лежит в основе его противоопухолевой активности и реализуется, в частности, через снижение лактацидоза и индукцию реактивных форм кислорода (ROS) [8-12]. Целью нашего исследования был анализ влияния лактацидоза на выживаемость клеток LLC/R9 в условиях дефицита питательных веществ in vitro и оценка противоопухолевой и антиметастатической активности DCA против LLC/R9 in vivo.
МАТЕРИАЛЫ И МЕТОДЫ
Экспериментальные животные, опухолевые клетки
В исследовании использовали 2,0-2,5-месячных мышей линии C57Bl/6 весом 18-23 г, выращенных на базе Института экспериментальной патологии, онкологии и радиобиологии имени Р.Е. Кавецкого НАН Украины. Протоколы исследований и процедуры работы с животными проводились в соответствии с основными требованиями к содержанию и работе с лабораторными животными и правилами местного комитета по биоэтике.
В исследовании использовали вариант LLC LLC/R9, полученный из штамма LLC дикого типа путем 9 последовательных сеансов химиотерапии in vivo на основе цис-диамминдихлороплатина (цис-ДДП) [13]. Клетки LLC/R9 поддерживали в культуральной среде RPMI (Sigma, США), дополненной 10% фетальной телячьей сывороткой (FCS) (Sigma, США), и 40 мг/мл гентамицина при 37 °C в увлажненной атмосфере с 5%CO2.
Эксперименты in vitro
Количество клеток в суспензии и их жизнеспособность регулярно анализировали на гемоцитометре с использованием теста исключения трипанового синего.
Для оценки влияния лактацидоза на жизнеспособность клеток LLC/R9, 1,5-105 клеток/лунку высевали в 24-луночный планшет в среду RPMI 1640 (Sigma, США) со стандартным содержанием глюкозы. После ночной инкубации клеток инкубационную среду заменяли свежей средой с различным содержанием глюкозы, лактата и с различным pH для моделирования условий дефицита глюкозы, лактацидоза на фоне дефицита глюкозы, а также стандартной (табл. 1). Глюкозодефицитную среду готовили на основе среды RPMI 1640 без глюкозы (Sigma, США). Лактацидоз генерировали путем добавления чистой молочной кислоты (Sigma, США) в среду с дефицитом глюкозы до конечной концентрации 14 ± 0,7 мМ и рН 6,7.
| Среда | Содержание глюкозы, мМ |
Содержание лактата, мМ | рН |
|---|---|---|---|
| Стандарт | 9.0 ± 0.5 | 1.6 ± 0.1 | 7.4 ± 0.01 |
| Дефицит глюкозы | 3.0 ± 0.1 | 1.6 ± 0.1 | 7.4 ± 0.01 |
| Лактацидоз | 3.0 ± 0.1 | 14.0 ± 0.7 | 6.7 ± 0.01 |
Таблица 1. Содержание глюкозы, лактата и рН культуральных сред, использованных в исследовании
Влияние различных условий инкубации на выживаемость опухолевых клеток, производство ROS, потребление глюкозы и производство лактата оценивали на2-й день инкубации опухолевых клеток.
Содержание глюкозы в культуральной среде и в гомогенатах опухолевой ткани определяли ферментным глюкозооксидантным методом с использованием набора для анализа глюкозы в биологических жидкостях (Sigma, США) согласно инструкции производителя. Содержание лактата в инкубационной среде и в гомогенатах опухолевой ткани определяли методом ферментной спектрофотометрии с использованием лактатдегидрогеназы (Sigma, США) [14]. Образцы среды и опухолевых тканей собирали и хранили при -20 °С или в жидком азоте, соответственно, до проведения измерений.
Уровень апоптоза в опухолевых клетках анализировали с использованием красителя Hoechst 33258 (Sigma, США) и флуоресцентной микроскопии по стандартной методике.
Продукцию ROS в опухолевых клетках определяли с использованием 2,7-дихлорфлуоресцеин диацетата (Sigma, США) методом спектрофлуорометрии (возбуждение при 495 нм, эмиссия при 530 нм) согласно [15].
Все измерения были повторены.
Эксперименты in vivo
Для экспериментов in vivo клетки LLC/R9 размножали in vitro в стандартных условиях и инокулировали i.m. мышам (1,0-106 клеток/животное в 0,1 мл раствора Хенкса).
После инокуляции клеток LLC/R9 животных распределили на 2 группы: группа 1 — мыши, обработанные DCA (Sigma, США) в общей дозе 1,5 г/кг (LD50/3) (n = 13); группа 2 — мыши, обработанные водой в том же режиме и в том же объеме (контроль, n = 12).
Лечение было начато на следующий день после трансплантации опухолевых клеток по метрономическому режиму, 5 раз в неделю в течение 3 недель. DCA готовили ex tempore в воде и вводили per os в объеме 0,4 мл/животное.
Объем первичной опухоли рассчитывали на основании ее диаметра, измеряемого штангенциркулем каждый3-й день, начиная с10-го дня после инокуляции опухолевых клеток, по формуле:
V = π (d)3/6,
где d — диаметр опухоли (мм).
Уровень метастазирования у мышей, несущих опухоль, оценивали на 21-й день после инокуляции опухолевых клеток по количеству и объему метастазов в легких с помощью бинокулярного микроскопа и миллиметровой шкалы.
Общий объем метастазов рассчитывали по формуле:
V = Σ niπ(di)3/6,
где V — общий объем метастазов (мм3), ni — количество метастазов с диаметромdi (мм).
Анализ функциональной активности компонентов дыхательной цепи митохондрий в опухолевых клетках проводили с использованием ЭПР на21-е сутки после инокуляции опухолевых клеток. Опухолевую ткань нарезали на образцы специального размера (d = 4,0 мм, l = 25-35 мм), замораживали и хранили при температуре -70 °С. ЭПР-анализ образцов проводили при 77 К на спектрофотометре Е-109 Varian (США) при скорости развертки потенциала 500 Е/мин, амплитуде модуляции 1,25×10 Е, мощности СВЧ-облучения 10.0 мВт, постоянном сеансе работы аппарата 1,0 с. По данным спектров ЭПР определяли уровни комплексов гемового железа с NO (при gmed = 2,007) в белках ЭТЦ митохондрий и белков Fe-S кластера (при g = 1,94) в опухолевых клетках.
Статистический анализ полученных результатов проводили с помощью описательных методов, нелинейного регрессионного анализа и t-теста Стьюдента с использованием программ Microsoft Excel и Microcal Origin.
РЕЗУЛЬТАТЫ
Было показано, что лактацидоз в условиях дефицита глюкозы значительно способствовал выживанию клеток LLC/R9. Действительно, кинетика роста опухолевых клеток, инкубированных в условиях лактацидоза на фоне дефицита глюкозы, существенно не отличалась от таковой у клеток, инкубированных в среде со стандартным содержанием глюкозы, по крайней мере, в период их экспоненциального роста. В частности, количество жизнеспособных клеток на2-е сутки инкубации в условиях лактацидоза на фоне дефицита глюкозы было практически таким же, как и в случае инкубации клеток в среде со стандартным содержанием глюкозы. В то же время в обоих случаях (лактацидоз и стандарт) количество жизнеспособных клеток было почти на 30% (р < 0,05) выше, чем при инкубации клеток в условиях дефицита глюкозы (рис. 1, а).
Кроме того, количество апоптотических клеток в условиях лактацидоза также статистически не отличалось от показателя в случае инкубации клеток в среде со стандартным содержанием глюкозы и на2-е сутки было равно 8,5 ± 0,9%, тогда как в условиях дефицита глюкозы количество апоптотических клеток было почти в три раза выше (р < 0,05), чем в случае лактацидоза (рис. 1, б).
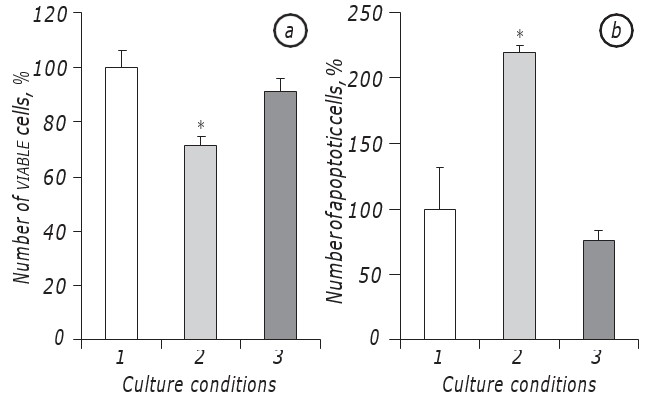 Рис. 1. Выживаемость клеток LLC/R9 на2-й день инкубации в стандартной (1), глюкозодефицитной (2) и лактацидозной (3) средах; а — количество жизнеспособных клеток; б — уровень апоптоза. *p < 0,05 по сравнению с контролем
Рис. 1. Выживаемость клеток LLC/R9 на2-й день инкубации в стандартной (1), глюкозодефицитной (2) и лактацидозной (3) средах; а — количество жизнеспособных клеток; б — уровень апоптоза. *p < 0,05 по сравнению с контролем
Интересно, что в условиях лактацидоза в клетках LLC/R9 потребление глюкозы было значительно ниже. Низкая скорость потребления глюкозы опухолевыми клетками при лактацидозе, зарегистрированная только на1-е сутки инкубации, восстанавливалась на2-е сутки и была на 70% ниже (p < 0,05), чем в случае среды со стандартным содержанием глюкозы (табл. 2). В случае дефицита глюкозы в противоположность лактацидозу на2-е сутки уровень глюкозы в инкубационной среде падал до нуля, что дополнительно свидетельствовало о снижении потребления глюкозы клетками LLC/R9 в условиях лактацидоза.
В то время как лактацидоз приводил к снижению скорости потребления глюкозы клетками LLC/R9, уровень внутриклеточных ROS в клетках, выживших в таких условиях, значительно возрастал. Эти данные представлены в таблице 2 и показывают, что уровень ROS в клетках, инкубированных в условиях лактацидоза, был почти на 150% (p < 0,05) и 230% (p < 0,05) выше соответствующих показателей для клеток, инкубированных в стандартной и глюкозодефицитной среде, соответственно.
Таким образом, полученные данные показали, что лактацидоз значительно способствовал выживанию клеток LLC/R9 в условиях дефицита глюкозы in vitro, что подтверждается высоким числом выживших в таких неблагоприятных условиях клеток и низким уровнем апоптоза. Выживание клеток было связано с неожиданным повышением уровня внутриклеточного ROS и снижением потребления глюкозы в LLC/R9.
| Среда | Потребление глюкозы, % | ROS, % |
|---|---|---|
| Стандарт | 100.0 ± 5.9 | 100.0 ± 24.8 |
| Дефицит глюкозы | 0.0 ± 0.0* | 75.8 ± 10.7 |
| Лактацидоз | 29.8 ± 1.5* | 248.7 ± 53.2* |
Таблица 2. Влияние лактацидоза в условиях дефицита глюкозы на потребление глюкозы и продукцию ROS опухолевыми клетками in vitro.
Примечание: *p < 0,05.
Данные закономерности выживания клеток LLC/R9 в условиях лактацидоза на фоне дефицита глюкозы in vitro свидетельствовали о том, что снижение содержания лактата в опухолевом микроокружении может препятствовать выживанию опухолевых клеток в условиях метаболического стресса, оказывая противоопухолевый эффект. Эта гипотеза была проверена нами с использованием DCA как соединения, способного уменьшать лактацидоз.
Данные о влиянии DCA на кинетику роста и метастазирование LLC/R9 представлены на рис. 2 и в табл. 3. Согласно этим данным, DCA не оказывал существенного влияния на рост первичных опухолей, но вызывал выраженное подавление метастазирования. Кинетика роста первичных опухолей у мышей с LLC/R9, обработанных ДКА, практически не отличалась от таковой у контрольных мышей, и на21-й день после пересадки опухоли объем первичных опухолей в опытной группе был всего на 39% меньше, чем в контрольной (см. рис. 2, табл. 3). Несмотря на то, что DCA не подавлял рост первичной опухоли, его антиметастатическая активность в отношении LLC/R9 оказалась поразительной. Количество и объем метастазов в легких у мышей с опухолью, получавших DCA, были на 59% (р < 0,05) и 94% (р < 0,05) ниже, чем эти показатели в контрольной группе, соответственно (см. табл. 3).
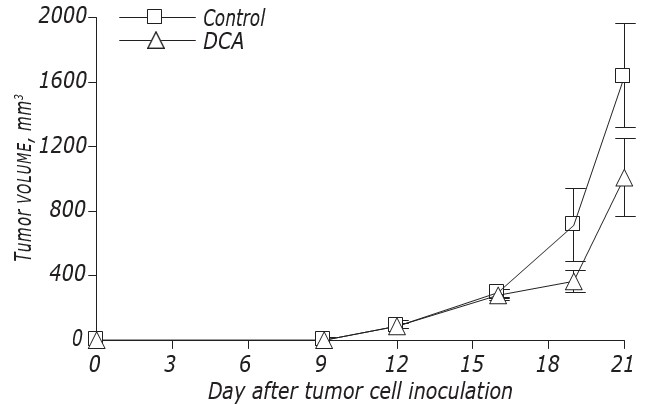 Рис. 2. Влияние DCA на кинетику роста LLC/R9 in vivo
Рис. 2. Влияние DCA на кинетику роста LLC/R9 in vivo
| Группа мышей | Объем опухоли, мм3 | Количество метастазов | Объем метастазов, мм3 |
|---|---|---|---|
| Контроль (n = 13) | 1702.7 ± 333.9 | 10.9 ± 1.2 | 17.9 ± 5.6 |
| ДКА (n = 13) | 1046.0 ± 258.3 | 4.5 ± 1.6* | 1.1 ± 0.4* |
Таблица 3. Влияние DCA на рост и метастазирование LLC/R9.
Примечание: *р < 0,05, различия значительны по сравнению со значением для контроля.
Анализ содержания лактата в образцах опухолевой ткани показал, что неожиданно DCA вызвал значительное увеличение содержания лактата в опухолевой ткани, по крайней мере, на21-й день после пересадки опухоли. Как видно из таблицы 4, содержание лактата в опухолевой ткани мышей, получавших DCA, было почти на 30% выше (р < 0,05), чем в контроле. Учитывая способность DCA как ингибитора PDH-киназы реорганизовывать энергетический метаболизм злокачественной опухоли в сторону окислительного фосфорилирования, мы рассматривали продукцию лактата опухолевыми клетками как суррогатный маркер ингибирования гликолиза при воздействии DCA. Повышение уровня лактата в опухоли под действием DCA показало, что его введение мышам с LLC/R9 в общей дозе 1,5 г/кг массы тела животного может быть недостаточным для активации окислительного фосфорилирования в опухолевых клетках, что отчасти объясняет его низкую эффективность против первичных опухолей.
| Группа мышей | Лактат (мкмоль/1 г ткани) |
|---|---|
| Контроль (n = 4) | 11.1 ± 0.6 |
| ДКА (n = 5) | 14.4 ± 1.5* |
Таблица 4. Влияние DCA на уровень лактата в опухолевой ткани мышей-носителей LLC/R9.
Примечание: р < 0,05, различия значительны по сравнению со значением для контроля.
Анализ спектров ЭПР опухолевых образцов показал, что ДКА не оказывал существенного влияния на функциональное состояние компонентов ЭТЦ в митохондриях опухолевых клеток (табл. 5). Например, у мышей с LLC/R9, леченных ДКА, интенсивность сигналов ЭПР, соответствующих белковым комплексам нитрозил-гемовое железо (gсер = 2,007) в белках ЭТК митохондрий опухолевых клеток, не была существенно выше, чем у контрольных мышей. Известно, что накопление NO-комплексов гемового железа может свидетельствовать, с одной стороны, о редокс-дисбалансе в сторону доминирования свободнорадикальных процессов, в частности, гиперпродукции NO, а с другой — о возможном ингибировании клеточного дыхания через нитрозилирование гемовых белков. Однако DCA, основной механизм противоопухолевого действия которого, как считается, связан с индукцией продукции ROS митохондриями [8, 10, 11], не вызывал повышения уровня комплексов железа гема с NO в опухолевой ткани. Последнее наблюдение, возможно, связано с особенностями клеток LLC/R9, а именно с чрезвычайно высоким содержанием этих комплексов, характерным для этой опухоли, прогрессивное накопление которых в процессе развития опухоли in vivo было зарегистрировано нами даже в отсутствие лечения [16].
| Относительная интенсивность сигнала ЭПР | Относительная интенсивность сигнала ЭПР | |
| Группа мышей | Нитрозил-гемовые белковые комплексы железа (g = 2,007) | Fe-S белок (g = 1.94) |
| Контроль | 54.3 ± 4.5 | 15.8 ± 0.5 |
| DCA | 97.8 ± 30.1 | 17.8 ± 2.1 |
Таблица 5. Влияние DCA на активность митохондриального ЭТЦ опухолевых клеток
Отсутствие существенного влияния ДКА на функциональную активность компонентов митохондриального ЭТЦ в опухолевых клетках подтверждается также данными об интенсивности сигналов ЭПР, соответствующих белкам Fe-S-кластера (g = 1,94) (комплексы І, ІІ, ІІІ), которая была практически одинаковой в обеих группах животных (см. табл. 5).
В заключение, результаты нашего исследования показали, что лактацидоз значительно способствовал выживанию варианта LLC LLC/R9 в условиях дефицита глюкозы. В то же время при развитии LLC/R9 in vivo DCA не оказывал противоопухолевой активности против первичных опухолей. Отсутствие противоопухолевого действия DCA против роста LLC/R9 согласуется с отсутствием ингибирующего эффекта DCA на содержание лактата в опухоли, а также с отсутствием заметного влияния DCA на продукцию ROS опухолевыми клетками. Хотя DCA не влиял на рост LLC/R9, но резко подавлял метастазирование, это наблюдение не может быть объяснено действием DCA в первичной опухоли, и требуются дальнейшие дополнительные исследования его антиметастатического действия.
ССЫЛКИ
1 Feron O. Пируват в лактат и обратно: от эффекта Варбурга до симбиотического обмена энергетического топлива в раковых клетках. Radiother Oncol 2009; 92: 329-33. doi: 10.1016/j.radonc.2009.06.025.
2 Wu H, Ding Z, Hu D, et al. Центральная роль молочнокислого ацидоза в устойчивости раковых клеток к вызванной недостатком глюкозы клеточной гибели. J Pathol 2012; 227: 189-99. doi: 10.1002/path.3978.
3 Fiaschi T, Marini A, Giannoni E, et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res 2012; 72: 5130-40.
4 Соляник Г.И., Федорчук А.Г., Пясковская О.Н., и др. Противораковая активность аконитинсодержащего растительного экстракта BC1. Exp Oncol 2004; 26: 307-11.
5 Пясковская О.Н. Антиангиогенное действие циклофосфамида на экспериментальные метастатические опухоли. J Med Chem 2012; 2: 25-9 (на украинском языке).
6 Колесник Д. Л., Пясковская О. Н., Трегубова Н. В., Соляник Г. И. Вариант карциномы легких Льюиса с высокой чувствительностью к противоопухолевой антиангиогенной терапии проявляет высокую способность к аутофагии. Cytol Genet 2012; 46: 155-60. doi: 10.3103/S009545271203005X.
7 Stacpoole PW. Фармакология дихлорацетата. Metabolism 1989; 38: 1124-44.
8 Bonnet S, Archer SL, Allalunis-Turner J, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007; 11: 37-51.
9 Wong JY, Huggins GS, Debidda M, et al. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol 2008; 109: 394-402. doi: 10.1016/j.ygyno.2008.01.038.
10 Michelakis ED, Sutendra G, Dromparis P, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2010; 2: 31-4. doi: 10.1126/scitranslmed.3000677.
11 Stockwin LH, Yu SX, Borgel S, et al. Sodium dichloroacetate selectively target cells with defects in the mitochondrial ETC Int J Cancer 2010; 127; 2510-19.
12 Kumar A, Kant S, Singh SM. Novel molecular mechanisms of antitumor action of dichloroacetate against T cell lymphoma: Влияние измененного метаболизма глюкозы, гомеостаза рН и регуляции выживания клеток. Chem Biol Interact 2012; 199: 29-37.
13 Pyaskovskaya ON, Dasyukevich OI, Kolesnik DL, et al. Changes in VEGF level and tumor growth characteristics during Lewis lung carcinoma progression towards cis-DDP resistance. Exp Oncol 2007; 29: 197-202.
14 Биохимические методы (липидный и энергетический обмен). М.И. Прохорова, ред. Л.: Ленинградский университет, 1982. 272 p.
15 Wang H, Joseph JA. Количественная оценка клеточного окислительного стресса с помощью анализа на дихлорфлуоресцеин с использованием микропланшетного ридера. Free Radic Biol Med 1999; 27: 612-6.
16 Пясковская О.Н., Сорокина Л.В., Колесник Д.Л., и др. Динамика изменений показателей антиоксидантной системы в процессе роста двух вариантов карциномы легких Льюиса. Exp Oncol 2014; 36: 29-33.