DCA усиливает противоопухолевое действие капецитабина в аллотрансплантате меланомы B16 мыши и ксенотрансплантате немелкоклеточного рака легкого A549 человека
DCA усиливает противоопухолевое действие капецитабина в аллотрансплантате меланомы B16 мыши и ксенотрансплантате немелкоклеточного рака легкого A549 человека
Оригинал статьи: https://www.sci-hub.ru/10.1007/s00280-013-2281-z
Кафедрабиохимии, факультет наук о жизни, Фуданьский университет, Хандан Роуд 220, Шанхай, 200433, Китай.
Электронная почта: e-mail: whuang@fudan.edu.cn
Получено: 16 мая 2013 г.
Принято: 27 августа 2013 г.
Опубликовано: 17 сентября 2013 г
Аннотация
Цель: Капецитабин является одним из немногих химиотерапевтических препаратов с высокой пероральной доступностью. Недавно дихлорацетат натрия (ДХА) показал большой потенциал в качестве противоракового агента. В настоящем исследовании мы оценили противораковый эффект DCA в комбинации с капецитабином при раке со скромной экспрессией TP.
Методы: Для оценки эффекта комбинированного лечения DCA и капецитабином использовали аллотрансплантат меланомы мыши B16 и ксенотрансплантат немелкоклеточного рака легкого человека A549. Гистология и иммуногистохимия использовались для выявления апоптоза и пролиферации раковых клеток. ПЦР в реальном времени и Вестерн-блот проводились для определения экспрессии TP и каспаз, соответственно.
Результаты: Впервые мы сообщаем, что DCA усиливает противоопухолевый эффект капецитабина в мышином аллотрансплантате B16 и человеческом ксенотрансплантате A549, способствуя апоптозу опухолевых клеток. DCA оказывает незначительное влияние на экспрессию TP.
Выводы: Наши результаты свидетельствуют о том, что DCA в комбинации с капецитабином может стать потенциально новым терапевтическим режимом против некоторых видов рака.
Ключевые слова: DCA, капецитабин, комбинация, противоопухолевый эффект
ВВЕДЕНИЕ
Дихлорацетат натрия (ДХА) — это маленькая молекулярная соль дихлоруксусной кислоты с молекулярной массой 150 Да. DCA ингибирует активность киназы пируватдегидрогеназы, тем самым активируя митохондриальный ферментный комплекс пируватдегидрогеназы [1] и переводя гликолитический путь метаболизма на окислительное фосфорилирование. В течение последних 40 лет DCA использовался в качестве сиротского препарата для лечения врожденного молочнокислого ацидоза у детей и молочнокислого ацидоза, осложненного другими заболеваниями [2], и показал высокую эффективность и низкую токсичность как в доклинических, так и в клинических испытаниях [3]. Недавно DCA продемонстрировал большой потенциал в качестве противоракового средства из-за сходства метаболического ремоделирования некоторых опухолевых клеток с процессами, происходящими при молочнокислом ацидозе [4]. Раковые клетки, особенно раковые стволовые клетки (РСК), сопротивляются апоптозу, производя энергию путем гликолиза и молочнокислого брожения, а не окислительного фосфорилирования, из-за гипоксической природы опухолевой микросреды — явления, известного как эффект Варбурга [5,6]. Было показано, что после перорального приема DCA восстанавливает функцию митохондрий и избирательно способствует апоптозу опухолевых клеток по митохондриально-зависимому пути [7,8]. Терапевтическая активность DCA против глиобластомы была проверена в клинических испытаниях (NCT00540176) и показала некоторые положительные результаты [9]. Однако исследование II фазы NCT01029925 по определению частоты ответа на пероральный прием дихлорацетата у пациентов с рецидивирующим и/или метастатическим и предварительно леченным раком молочной железы и немелкоклеточным раком легкого было прекращено из-за более высокого, чем ожидалось, риска и проблем с безопасностью. Таким образом, клиническая польза DCA для борьбы с раком нуждается в более тщательной оценке.
Как сенсибилизатор апоптоза, DCA также использовался в комбинации с другими методами лечения рака. Cao и другие [10] сообщили, что DCA сенсибилизировал клетки рака простаты к радиации in vitro. Сяо и др. [11] установили, что DCA усиливает гибель опухолевых клеток в сочетании с онколитическим аденовирусом, экспрессирующим опухолевый супрессор MDA-7/IL-24. Недавно метаболическая таргетная терапия с использованием DCA была продемонстрирована как новая стратегия лечения для улучшения результатов фотодинамической терапии [12]. Тонг и др. [13] обнаружили, что DCA и 5-фторурацил проявляют синергетический противоопухолевый эффект в клетках колоректального рака in vitro. Однако до сих пор существуют противоречивые результаты и сомнения относительно применения DCA отдельно или в комбинации с другими препаратами. Шахрзад и др. [14] показали, что DCA снижает апоптоз раковых клеток в гипоксических условиях как in vitro, так и in vivo. Хеше и др. [15] предупредили, что DCA снижает цитотоксичность некоторых стандартных противораковых препаратов, таких как цисплатин и доксорубицин, но не влияет на активность темозоломида в 7 из 10 клеточных линий в их исследовании. Эти противоречивые результаты означают, что применение DCA отдельно или в комбинации с другими методами лечения может зависеть от типа рака и конкретного агента.
Капецитабин является одним из немногих химиотерапевтических препаратов с высокой пероральной доступностью и лицензирован в качестве первой линии лечения метастатического рака прямой кишки или альтернативного лечения метастатического рака молочной железы в сочетании с доцетакселом [16,17]. Капецитабин является пролекарством 5-фторурацила (5-ФУ) и требует 3 ферментативных реакций для окончательного превращения в 5-ФУ в опухолевых клетках. Последняя реакция катализируется тимидинфосфорилазой (TP), которая в некоторых опухолях выражена в большей степени, чем в нормальных тканях [18]. Поэтому цитотоксический 5-ФУ образуется в большей степени в опухолевых клетках, чем в тканях вне опухоли, что делает капецитабин малотоксичным химиотерапевтическим препаратом [18]. Уровни экспрессии TP различны в разных типах опухолей [18]; это ограничивает применение капецитабина только несколькими типами рака. В настоящем исследовании мы оценили противораковый эффект DCA в комбинации с капецитабином для раковых опухолей со скромной экспрессией TP. Мы предположили, что DCA усиливает противораковый эффект и снижает эффективную дозу капецитабина. Комбинация DCA с капецитабином может дать хорошую схему лечения, поскольку оба агента можно принимать перорально при хорошей приверженности пациентов. Кроме того, генерические формы DCA могут снизить эффективную дозу капецитабина, тем самым уменьшая побочные эффекты и стоимость лечения рака.
Материалы и методы
Материалы
Дихлорацетат натрия (ДХА, CSA:2156-56-1), чистота 99 %, был получен от компании Shanghai Jieshi Chemical Co. (Китай). 5-фторурацил (5-FU) и 5′-дезокси-фторуридин (5DFUR) были получены от Sigma-Aldrich (США). МТТ был получен от Shanghai Biological Engineering Co. (Китай). Таблетки капецитабина (Xeloda) были получены от Roche (США). Мышиная меланома B16 и человеческая немелкоклеточная линия клеток рака легкого A549 были получены из Американской коллекции клеточных культур (ATCC, США).
Исследования на животных моделях
Модель аллотрансплантата
Мыши C57BL/6, самки, возраст 6-8 недель, весом около 18-20 г, были приобретены в Шанхайском центре лабораторных животных (SLAC, Китай) и акклиматизированы в течение 1 недели. Один миллион одиночных клеток B16 был подкожно (s.c.) инокулирован в правый фланг мышей C57BL/6. Мышей разбили на случайные группы, по 6 мышей на группу в клетке. Было две группы мышей. DCA и капецитабин вводили этим двум группам мышей через 3 и 10 дней после инокуляции, соответственно. DCA добавляли в стерильную питьевую воду до конечной концентрации 1,4 г/л. Измерение объема потребленной воды показало, что количество ДКА, введенного каждой мыши, было приблизительно равно 100 мг/кг/день. Таблетки капецитабина измельчали и суспендировали в стерильной воде с 4 % карбоксиметилцеллюлозы для получения различных концентраций. Двести микролитров суспензии капецитабина вводили внутрижелудочно каждой мыши. Каждые 2 дня длинный (a) и короткий (b) диаметры опухолей измеряли с помощью штангенциркуля, а также регистрировали массу тела. Объем опухоли рассчитывали по формуле V = 0,5ab2. Через 22 дня после инокуляции мышей умертвили, опухоли удалили и взвесили.
Модель ксенотрансплантата
Мыши BALB/c-nu, самцы, возраст 5-6 недель, весом около 18-20 г, были приобретены в Шанхайском центре лабораторных животных (SLAC, Китай) и акклиматизированы в течение 1 недели. Приблизительно 2 × 2 мм срезы только что измельченных опухолевых тканей A549, полученных от мышей BALB/c-nu, ранее инокулированных клетками A549, были введены внутривенно в область правого фланга самцов мышей BALB/c-nu. DCA и капецитабин вводили мышам, когда объем опухоли достигал ~0,2 см3. Через тридцать-тридцать пять дней после лечения мышей умерщвляли, опухоли удаляли и взвешивали. Другие методы были такими же, как описанные в эксперименте с аллотрансплантацией.
Исследования на животных были одобрены группой по благополучию животных и этике факультета лабораторных животных Фуданьского университета.
Гистология и иммуногистохимия
Гистологическое исследование опухолевых узлов проводили с использованием дополнительных животных (по 3 мыши в каждой группе), которые не рассматривались для мониторинга роста опухоли. Ткани фиксировали в 4 % (w/v) параформальдегиде, после фиксации в течение ночи при комнатной температуре образцы обезвоживали в градуированном этаноле и встраивали в парафин. После этого различные части опухоли произвольно вырезали для получения 4-мкм срезов на микротоме Leica. После депарафинизации и регидратации три среза из разных частей каждого образца были отобраны для последующих операций. Терминальное дезоксинуклеотидилтрансфераза-опосредованное никелирование концевого участка ДУТФ (TUNEL) и окрашивание 4′6-диамидино-2-фенилиндолом (DAPI) проводили в соответствии с инструкциями производителей наборов TUNEL и DAPI (Beyotime, Китай). Срезы анализировали с помощью инвертированного флуоресцентного микроскопа (Olympus, Япония). Выявление ядерного антигена пролиферирующих клеток (PCNA) проводили после депарафинизации срезов и инкубации при 96-100 °C в течение 20 мин. Активность эндогенной пероксидазы гасили 0,3 % (v/v) перекисью водорода в 60 % (v/v) метаноле в течение 30 мин. Неспецифическую адсорбцию минимизировали путем инкубации срезов в 2 % (v/v) нормальной козьей сыворотке в PBS в течение 20 мин. Участки тканей инкубировали в течение ночи с кроличьим поликлональным антителом анти-PCNA (Abcam; 1:100 в PBS), промывали PBS 3 раза, по 30 мин каждый раз, и инкубировали с биотин-конъюгированным козьим антирабическим IgG в течение 2 ч при 37 °C и авидин-биотин-пероксидазным комплексом в течение 1 ч при 37 °C. Срезы контрастировали гематоксилином (Sigma-Aldrich) и анализировали с помощью световой микроскопии (Olympus, Япония). Для анализа иммуногистохимии использовали по три опухоли на группу. Один или два среза на опухоль были слепо отобраны для измерения PCNA- или TUNEL-позитивных клеток. Пять случайных полей на слайде при увеличении 400× измеряли вслепую (n = 250 клеток на группу). При проведении количественного анализа TUNEL-позитивных клеток, возможные некротические клетки исключались путем наблюдения за ядерной морфологией с помощью окрашивания DAPI. Для обеспечения точности результатов проводили как положительный, так и отрицательный контроль.
Выделение белка и вестерн-блоттинг
Опухолевые ткани из каждой группы (3 дополнительные мыши в каждой группе) были объединены вместе и измельчены под жидким азотом, а затем лизированы в 150
мкл
буфера для лизиса тканей (Beyotime, Китай). Пробирки энергично встряхивали в течение 1 мин, помещали на лед на 20 мин и центрифугировали при 5 000g в течение 5 мин при 4 °C. Концентрацию общего белка определяли с помощью набора для определения белка BCA (BioRad). Тридцать микрограммов общего белка из каждого образца разделяли методом SDS-PAGE на 10 % геле, переносили на PVDF мембрану, блокировали, инкубировали в течение ночи с первичным антителом, инкубировали в течение 1 ч с вторичным антителом и окрашивали хромогенным субстратом NBT/BCIP. Мышиное моноклональное антитело против антикаспазы 3 (1:500), антитело против антикаспазы 9 (1:1 000), анти-β-актин (1:2 000) и кроличье поликлональное антитело против антикаспазы 8 (1:1 000) были получены от Beyotime (Нанкин, Китай). Мышиное моноклональное антитело анти-ТФ (1:2,000) было получено от Abcam (Великобритания). в качестве внутреннего контроля использовали β-актин. Плотность полос вестерн-блота анализировали с помощью программы Clinx Gel Analysis V2.02 (Clinx Science, Китай).
ПЦР в реальном времени
Опухоли B16 и ткани печени были взяты от одной и той же мыши C57BL/6, ранее инокулированной клетками B16 (использовали 3 мышей, подарок Wenlong Ren из Шанхайского института фармацевтической промышленности). Colo205/A549 и ткани печени были взяты от одной и той же мыши BALB/c-nu, ранее инокулированной клетками Colo205/A549 (3 мыши были использованы, соответственно, подарок Wenlong Ren из Шанхайского института фармацевтической промышленности). Для анализа экспрессии TP после лечения образцы опухолей объединяли из 3 опухолевых тканей одинакового веса в каждой группе. Тотальную РНК выделяли с помощью реагента Trizol (Invitrogen, США) и подвергали обратной транскрипции с помощью набора реагентов PrimeScript® RT (Takara, Япония). кДНК нормировали на β-актин. ПЦР в реальном времени проводили трехэтапным методом с использованием набора SYBR® Premix Ex Taq™ II (Takata, Япония) с температурой отжига 55 °C и 40 циклами амплификации. Отдельный тест проводили в трех экземплярах. в качестве внутреннего контроля использовали β-актин. Относительное количество каждой кДНК анализировали с помощью2-△△Ct. Праймеры для ПЦР в реальном времени: β-актин F: 5′-TCAAGATCATTGCTC CTCCTG-3′ и β-актин R: 5′-CTGCTTGCTGATCCACATCTG-3′, hTP F: 5′-TGGCTCAGTCGGGACAGCAG-3′ и hTP R: 5′-TCCGCTGATCATTG GCACCT-3′, mTP F: 5′-GCCTAGCTAAAGCATTGTGCTC-3′ и mTP R: 5′-AAGGGTGCT CGATCTGATAGCA-3′.
Статистика
Мы использовали множественные сравнения ANOVA с post hoc анализом (тест Тьюки), используя программное обеспечение SPSS 16.0 (SPSS Inc., США). Данные представлены как среднее ± SEM, значимым считалось P < 0,05.
Результаты
Экспрессия TP в меланоме мыши B16 и опухолях человека A549 NSCLC
Экспрессия TP в опухолях B16 и A549, удаленных у мышей, была проанализирована методом ПЦР в реальном времени. Печень человека экспрессирует относительно больше TP, чем другие нормальные ткани [18]. В типичных типах рака, подходящих для лечения капецитабином, экспрессия TP в опухоли близка или выше, чем в печени, например, в колоректальном раке и раке молочной железы [18,19]. В ксенотрансплантатах рака человека раковые опухоли с высокой активностью TP были более восприимчивы к лечению капецитабином, чем раковые опухоли с низкой активностью TP [20]. Клеточная линия колоректального рака Colo205 была выбрана в качестве эталона на основании ее умеренной экспрессии TP и умеренной чувствительности к капецитабину [21]. Как показано на рис. 1a, уровень транскрипции TP в Colo205 был немного выше, чем в печени мыши BALB/c-nu. Как показано на рис. 1б, в, уровни транскрипции TP в опухолях B16 и A549 были близки к таковым в печени мышей. Таким образом, B16 и A549 также являются умеренно экспрессирующими TP клеточными линиями. Мы можем сделать вывод, что B16 и A549 будут в определенной степени реагировать на лечение капецитабином, не перекрывая эффект DCA. Таким образом, модели аллотрансплантата B16 и ксенотрансплантата A549 были пригодны для изучения противоопухолевого эффекта DCA и капецитабина в комбинации.
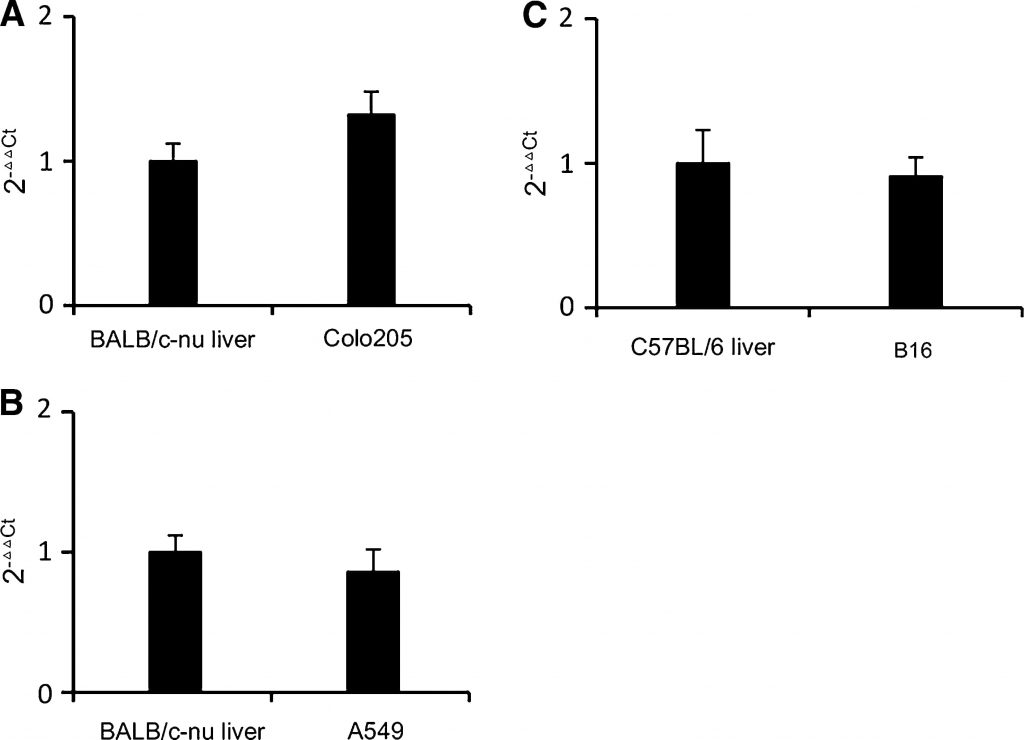 Рисунок 1. По сравнению с печенью мыши, опухоли B16 и A549 скромно экспрессируют TP ПЦР в реальном времени Анализ уровня транскрипции TP. Использовались праймеры, специфически нацеленные на ТП мыши и человека. Праймер на β-актин подходит как для мышей, так и для человека. a Уровень транскрипции TP в опухоли Colo205 по сравнению с уровнем транскрипции в печени мышей-носителей BALB/c-nu. b Уровень транскрипции TP в опухоли A549 по сравнению с уровнем транскрипции в печени мышей-носителей BALB/c-nu. c Уровень транскрипции TP в опухоли B16 по сравнению с уровнем транскрипции в печени мышей-носителей C57BL/6. В каждой группе использовались образцы от 3 мышей
Рисунок 1. По сравнению с печенью мыши, опухоли B16 и A549 скромно экспрессируют TP ПЦР в реальном времени Анализ уровня транскрипции TP. Использовались праймеры, специфически нацеленные на ТП мыши и человека. Праймер на β-актин подходит как для мышей, так и для человека. a Уровень транскрипции TP в опухоли Colo205 по сравнению с уровнем транскрипции в печени мышей-носителей BALB/c-nu. b Уровень транскрипции TP в опухоли A549 по сравнению с уровнем транскрипции в печени мышей-носителей BALB/c-nu. c Уровень транскрипции TP в опухоли B16 по сравнению с уровнем транскрипции в печени мышей-носителей C57BL/6. В каждой группе использовались образцы от 3 мышей
DCA усиливает противоопухолевый эффект капецитабина в аллотрансплантате мышиной меланомы B16 без дополнительной токсичности
Поскольку гипоксическая природа опухолевой микросреды является критической для оптимальной активности DCA, противоопухолевый эффект DCA плюс капецитабин был протестирован на животных моделях вместо клеточных линий. Тридцать шесть мышей C57BL/6 были инокулированы 1 ×106 клетками меланомы B16 и случайным образом разделены на 6 групп (n = 6): контрольная группа, не получавшая лекарств, группа, получавшая только DCA, группа, получавшая только капецитабин 10 мг/день, и три группы, получавшие DCA плюс капецитабин 5, 10 или 20 мг/день. Через три дня после инокуляции опухолевых клеток мышам вводили DCA в питьевой воде и перорально (p.o.) капецитабин в качестве отдельных агентов или в комбинации с возрастающими концентрациями капецитабина. Как показано на верхней панели рис. 2a, как DCA, так и капецитабин в дозе 10 мг/день в одиночку незначительно подавляли рост опухолей меланомы B16 по сравнению с контрольной группой. Напротив, DCA плюс 10 мг/день капецитабина значительно усиливали ингибирование роста опухоли (P < 0,05). Противоопухолевый эффект DCA плюс 20 мг/день капецитабина был аналогичен тому, который наблюдался при использовании DCA плюс 10 мг/день капецитабина; рост опухоли был почти полностью подавлен. Примечательно, что у мышей, получавших DCA плюс 20 мг/день капецитабина, наблюдалось резкое снижение массы тела (рис. 2а, нижняя панель), в то время как DCA плюс 10 мг/день капецитабина практически не влиял на массу тела по сравнению с контролем.
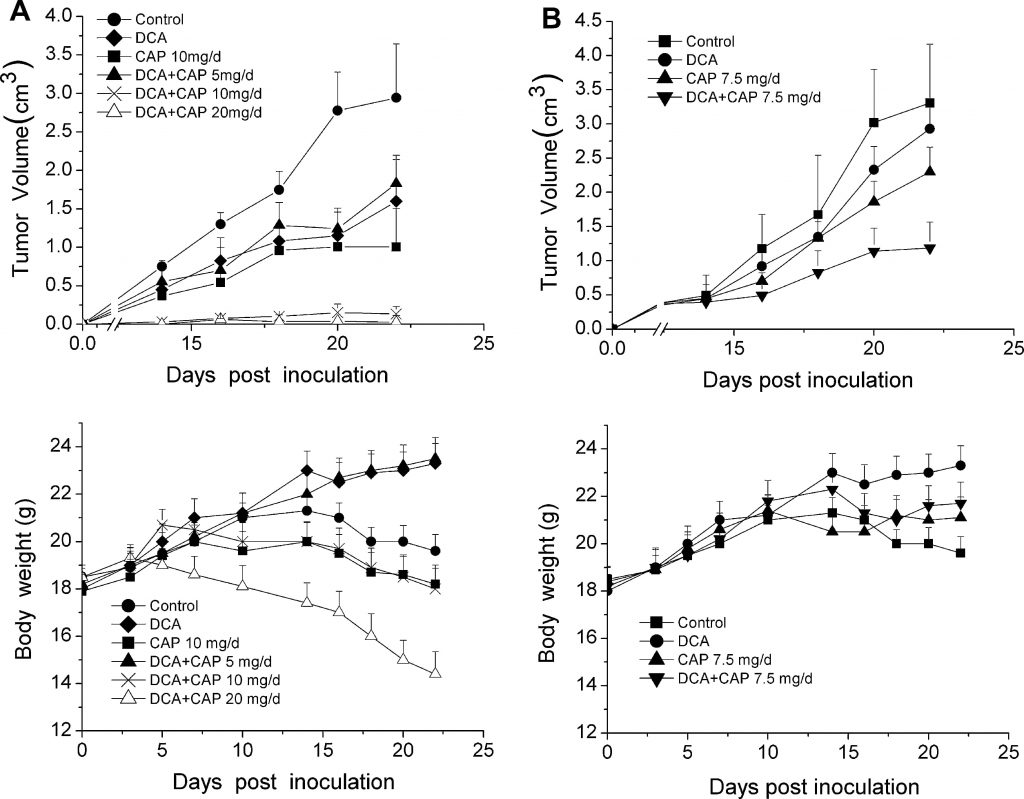 Рисунок 2. DCA усиливает противоопухолевый эффект капецитабина в аллотрансплантате меланомы B16 у мышей без дополнительной токсичности. a Через три дня после инокуляции мышам вводили DCA и капецитабин (CAP) по отдельности или в комбинации. Вводили эскалированные дозы [5, 10 и 20 мг/день (мг/день)] капецитабина в комбинации с постоянной дозой DCA. Показаны кривая объема опухоли(верхняя панель) и кривая массы тела(нижняя панель). b Через 10 дней после прививки мышам вводили DCA и 7,5 мг/день капецитабина отдельно или в комбинации. Представлены кривая объема опухоли(верхняя панель) и кривая массы тела(нижняя панель)
Рисунок 2. DCA усиливает противоопухолевый эффект капецитабина в аллотрансплантате меланомы B16 у мышей без дополнительной токсичности. a Через три дня после инокуляции мышам вводили DCA и капецитабин (CAP) по отдельности или в комбинации. Вводили эскалированные дозы [5, 10 и 20 мг/день (мг/день)] капецитабина в комбинации с постоянной дозой DCA. Показаны кривая объема опухоли(верхняя панель) и кривая массы тела(нижняя панель). b Через 10 дней после прививки мышам вводили DCA и 7,5 мг/день капецитабина отдельно или в комбинации. Представлены кривая объема опухоли(верхняя панель) и кривая массы тела(нижняя панель)
Для оценки противоопухолевого эффекта DCA плюс капецитабин против пальпируемых, обнаруживаемых опухолей, через 10 дней после инокуляции опухолевых клеток, DCA и капецитабин отдельно или в комбинации вводили второй группе из 36 мышей C57BL/6, инокулированных 1 ×106 клетками меланомы B16. Мыши с пальпируемыми опухолями были случайным образом разделены на 4 группы (n = 9, 3 мыши использовались для анализа иммуногистохимии): контроль, только DCA, только капецитабин в дозе 7,5 мг/день и DCA плюс капецитабин в дозе 7,5 мг/день. Как показано на верхней панели рис. 2б, DCA плюс капецитабин 7,5 мг/день значительно подавляли рост опухоли по сравнению с DCA или капецитабином (P < 0,05). Через 22 дня после инокуляции DCA плюс 7,5 мг/день капецитабина подавляли рост опухоли на 75 % (P < 0,05), в то время как только DCA и капецитабин подавляли рост только на 25 % и 35 %, соответственно (P < 0,05). DCA не вызывал резкой потери массы тела по сравнению с лечением только капецитабином (рис. 2b, нижняя панель). Эти результаты показывают, что DCA и капецитабин могут оказывать синергетический противоопухолевый эффект в опухолях меланомы B16.
DCA усиливает противоопухолевый эффект капецитабина в ксенотрансплантационной модели НСКЛ человека A549 без дополнительной токсичности
Ранее сообщалось, что НСКЛ человека можно лечить либо DCA [7], либо капецитабином [22-24]. В настоящем исследовании мы изучили противоопухолевый эффект DCA плюс капецитабин в модели ксенотрансплантата НСКЛ A549 человека. Шестьдесят шесть самцов мышей BALB/c-nu с опухолями человека NSCLC A549 (~2 × 2 мм), привитыми внутривенно в правый фланг, были случайным образом разделены на 9 групп: контроль; только DCA; 2,5, 5, 7,5 или 10 мг/день только капецитабина; или DCA плюс 2,5, 5 или 7.5 мг/день капецитабина (n = 6, кроме контроля, только DCA, 7,5 мг/день капецитабина и DCA плюс 7,5 мг/день капецитабина; в этих группах n = 9, 3 мыши были использованы для иммуногистохимического анализа и анализа Вестерн-блот или ПЦР в реальном времени). Капецитабин в дозе 10 мг/день был установлен в качестве контроля высокой дозы. Когда объем опухоли достигал 0,15-0,2 см3, мышам вводили препараты. Капецитабин вводили внутривенно по схеме 14 дней в день/7 дней в день. Как показано на левой панели рис. 3a, b и c, только DCA оказывал незначительное ингибирующее действие на опухоли A549; этот результат не согласуется с данными предыдущих отчетов [7], в которых только DCA проявлял больший противоопухолевый эффект. Только капецитабин в дозе 10 мг/день значительно подавлял рост опухолей A549, но при этом отмечалось резкое снижение массы тела, что указывает на сильную токсичность (рис. 3a, b, c, правые панели). Как показано на левой панели рис. 3а, 2,5 мг/день только капецитабина значительно уменьшали рост опухолей A549; комбинация DCA плюс 2,5 мг/день капецитабина усиливала ингибирование роста. Кривая увеличения объема опухоли при лечении только DCA предполагает, что капецитабин оказывает доминирующее противоопухолевое действие при комбинированном лечении. Эффект DCA плюс 5 мг/сут капецитабина был немного лучше, чем DCA плюс 2,5 мг/сут капецитабина, хотя и хуже, чем 10 мг/сут капецитабина, и не наблюдалось значительного снижения массы тела (рис. 3б, правая панель). Примечательно, что DCA плюс 5 мг/сут капецитабина значительно усилили ингибирование роста опухоли по сравнению с 5 мг/сут только капецитабина (рис. 3б, левая панель). Противоопухолевый эффект комбинации был близок к лечению капецитабином 10 мг/день, но без значительной потери массы тела (рис. 3б, правая панель). Эффект только капецитабина 7,5 мг/сут (рис. 3c) был немного лучше, чем капецитабина 5 мг/сут; однако при сочетании с ДКА не было значительной разницы между ДКА плюс капецитабин 7,5 мг/сут и ДКА плюс капецитабин 5 мг/сут. Эти результаты означают, что DCA может снизить дозу капецитабина без потери противоопухолевого эффекта или увеличения токсичности.
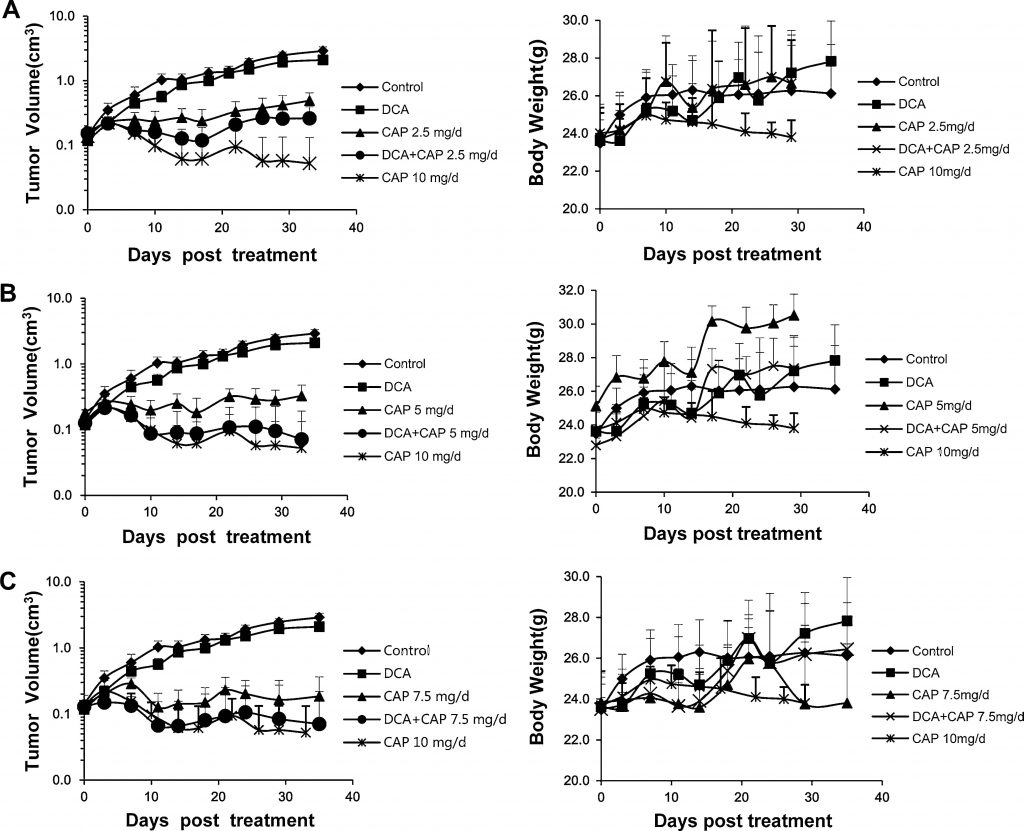 Рисунок 3. DCA увеличивает противоопухолевый эффект капецитабина в ксенотрансплантационной модели NSCLC A549 человека без дополнительной токсичности. a DCA плюс 2,5 мг/день (мг/день) капецитабина. b DCA плюс 5 мг/день капецитабина. c DCA плюс 7,5 мг/день капецитабина. Группа лечения капецитабином 10 мг/день использовалась в качестве контроля больших доз. Показаны кривые роста опухоли(левые панели) и кривые массы тела(правые панели). Объем опухоли представлен на логарифмической оси
Рисунок 3. DCA увеличивает противоопухолевый эффект капецитабина в ксенотрансплантационной модели NSCLC A549 человека без дополнительной токсичности. a DCA плюс 2,5 мг/день (мг/день) капецитабина. b DCA плюс 5 мг/день капецитабина. c DCA плюс 7,5 мг/день капецитабина. Группа лечения капецитабином 10 мг/день использовалась в качестве контроля больших доз. Показаны кривые роста опухоли(левые панели) и кривые массы тела(правые панели). Объем опухоли представлен на логарифмической оси
DCA усиливает апоптотический эффект капецитабина на клетки B16 и A549 in vivo
Гистологическое исследование опухолей меланомы B16 проводилось через 7 дней после начала лечения. Окрашивание TUNEL показало, что лечение опухолей меланомы B16 только DCA или 7,5 мг/день капецитабина индуцировало клеточный апоптоз на 8 и 17 %, соответственно. В комбинации DCA плюс 7,5 мг/день капецитабина индуцировали апоптоз примерно на 30 %, что больше, чем сумма отдельных препаратов вместе взятых (рис. 4a, c, левая панель). Окрашивание PCNA в опухолях меланомы B16 показало, что только DCA оказывал незначительное влияние на пролиферацию, в то время как капецитабин 7,5 мг/день значительно снижал пролиферацию. DCA плюс 7,5 мг/день капецитабина не усиливали ингибирование пролиферации только капецитабином в опухолевых клетках B16 (рис. 4b, c, правая панель).
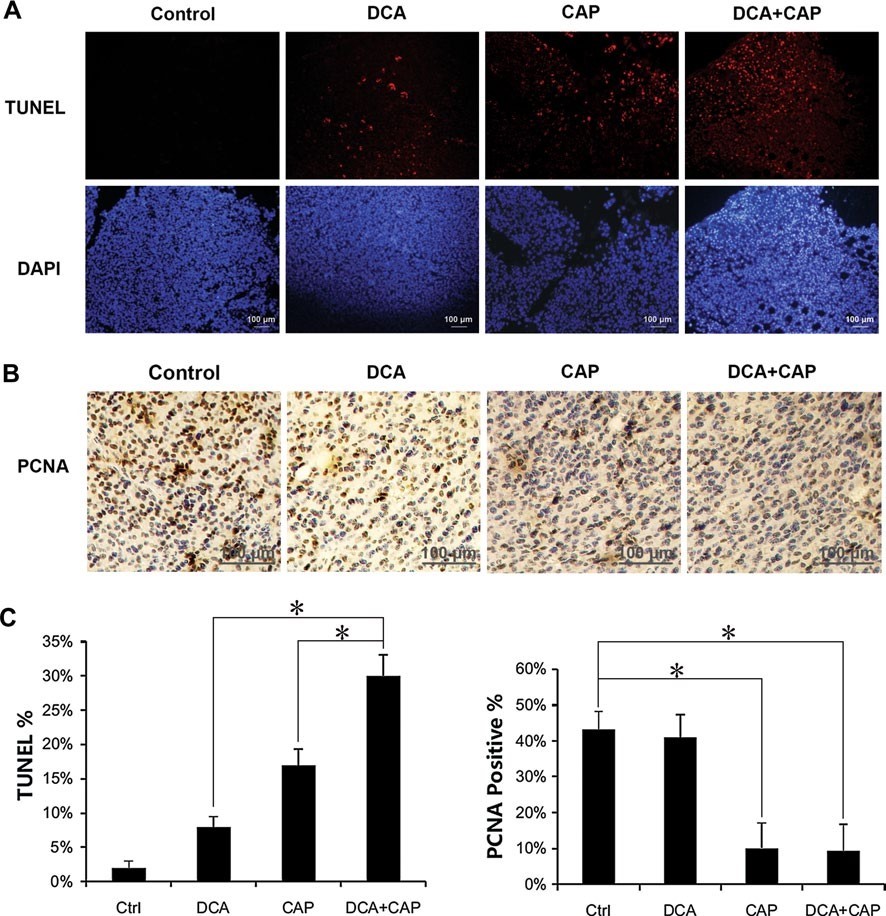 Рисунок 4. DCA усиливает апоптотический эффект капецитабина в опухолях меланомы B16. a Иммунофлуоресцентное окрашивание TUNEL образцов опухолей меланомы B16 от мышей, получавших DCA, 7,5 мг/день только капецитабина или DCA плюс 7,5 мг/день капецитабина в течение 7 дней. b Иммуногистохимический анализ антигена пролиферации PCNA в образцах опухолей меланомы B16 от мышей, получавших DCA, 7,5 мг/день капецитабина или DCA плюс 7,5 мг/день капецитабина в течение 7 дней. c Количественный анализ TUNEL-позитивных клеток(слева) и PCNA-позитивных клеток(справа). *P < 0.05
Рисунок 4. DCA усиливает апоптотический эффект капецитабина в опухолях меланомы B16. a Иммунофлуоресцентное окрашивание TUNEL образцов опухолей меланомы B16 от мышей, получавших DCA, 7,5 мг/день только капецитабина или DCA плюс 7,5 мг/день капецитабина в течение 7 дней. b Иммуногистохимический анализ антигена пролиферации PCNA в образцах опухолей меланомы B16 от мышей, получавших DCA, 7,5 мг/день капецитабина или DCA плюс 7,5 мг/день капецитабина в течение 7 дней. c Количественный анализ TUNEL-позитивных клеток(слева) и PCNA-позитивных клеток(справа). *P < 0.05
Подобно опухолям меланомы B16, лечение опухолей NSCLC A549 только DCA или 7,5 мг/день капецитабина индуцировало апоптоз на 15 и 30 %, соответственно. При совместном применении DCA плюс 7,5 мг/сут капецитабина индуцировали апоптоз на 50 %, что больше, чем сумма лечения одним агентом вместе взятым (рис. 5a, b). Эти результаты позволяют предположить, что DCA и капецитабин оказывают синергетическое действие на апоптоз опухолевых клеток NSCLC A549. DCA не усиливал ингибирование пролиферации капецитабином в опухолевых клетках NSCLC A549 (данные не показаны).
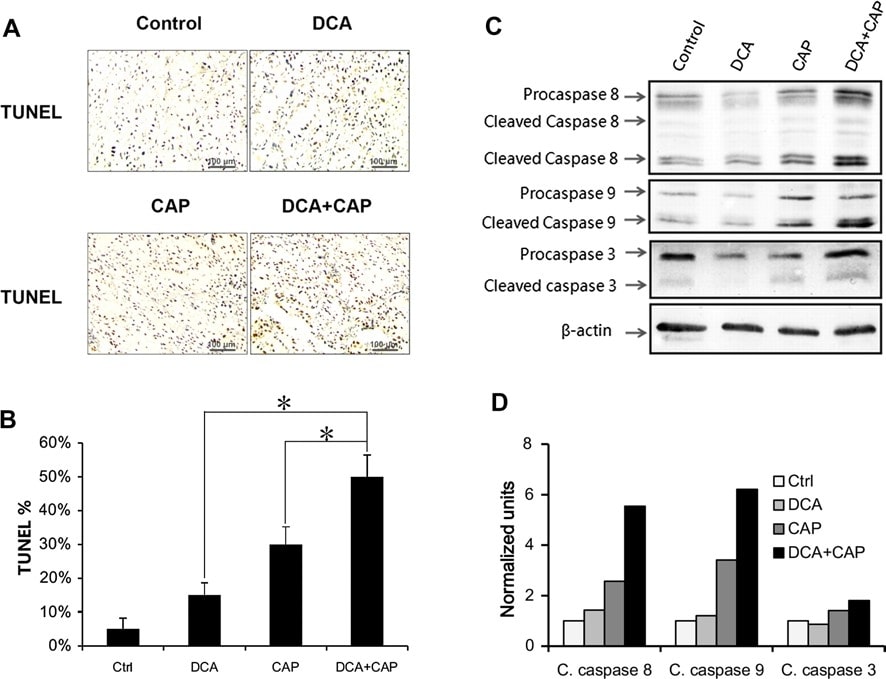 Рисунок 3. DCA усиливает апоптотический эффект капецитабина в опухолях NSCLC A549. a Иммуногистохимический анализ TUNEL образцов опухолей NSCLC A549 от мышей, получавших DCA, 7,5 мг/день только капецитабина или DCA плюс 7,5 мг/день капецитабина в течение 7 дней. b Количественный анализ TUNEL-позитивных клеток. c Вестерн-блот активации каспаз в опухолях NSCLC A549. Активация инициаторных каспаз (каспазы 8 и каспазы 9) и эффекторных каспаз (каспазы 3) была обнаружена в опухолях NSCLC A549, инокулированных мышам BALB/c-nu, получавшим DCA, 7,5 мг/день капецитабина или DCA плюс 7,5 мг/день капецитабина. d Плотный анализ расщепленных каспаз (C. caspase). Нормализовано по β-актину. *P < 0.05
Рисунок 3. DCA усиливает апоптотический эффект капецитабина в опухолях NSCLC A549. a Иммуногистохимический анализ TUNEL образцов опухолей NSCLC A549 от мышей, получавших DCA, 7,5 мг/день только капецитабина или DCA плюс 7,5 мг/день капецитабина в течение 7 дней. b Количественный анализ TUNEL-позитивных клеток. c Вестерн-блот активации каспаз в опухолях NSCLC A549. Активация инициаторных каспаз (каспазы 8 и каспазы 9) и эффекторных каспаз (каспазы 3) была обнаружена в опухолях NSCLC A549, инокулированных мышам BALB/c-nu, получавшим DCA, 7,5 мг/день капецитабина или DCA плюс 7,5 мг/день капецитабина. d Плотный анализ расщепленных каспаз (C. caspase). Нормализовано по β-актину. *P < 0.05
Вестерн-блот показал, что DCA не оказывает значительного влияния на экспрессию и активацию прокаспазы 8, прокаспазы 9 и прокаспазы 3 в опухолях NSCLC A549 по сравнению с контролем на 7-й день после лечения (рис. 5c, d). Только капецитабин в дозе 7,5 мг/день увеличивал активацию всех трех прокаспаз (рис. 5c, d). Интересно, что хотя DCA сам по себе не оказывал значительного влияния на экспрессию и активацию трех прокаспаз, DCA плюс капецитабин повышали экспрессию прокаспазы 8 и прокаспазы 3 по сравнению с лечением только капецитабином и усиливали активацию прокаспазы 8, прокаспазы 9 и прокаспазы 3.
DCA оказывает незначительное влияние на экспрессию TP в опухоли
Мы проанализировали экспрессию TP в образцах опухоли из ксенотрансплантата A549 на 7-й день после лечения. ПЦР в реальном времени (рис. 6a) и Вестерн-блот (рис. 6b) показали, что DCA как отдельный агент или в комбинации с капецитабином мало влияет на экспрессию TP, что означает, что механизм действия DCA и капецитабина в комбинации отличается от других синергистов капецитабина, о которых сообщалось ранее [19,25-27]. ПЦР в реальном времени также показала, что DCA не влияет на экспрессию других ферментов метаболизма капецитабина (тимидилат синтазы, оротат фосфорибозилтрансферазы, дигидропиримидин дегидрогеназы, тимидин киназы 1 и цитидин деаминазы, данные не показаны). Полученные результаты позволяют предположить, что DCA оказывает незначительное влияние на метаболизм капецитабина, что не приведет к повышению токсичности капецитабина.
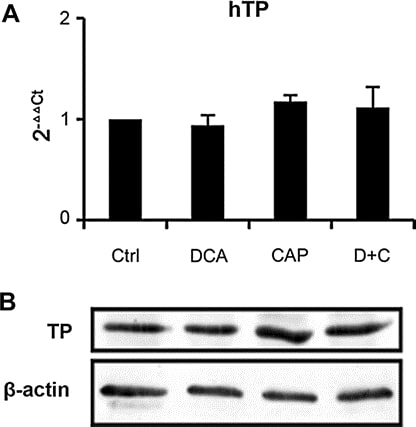 Рисунок 6. Транскрипция и экспрессия TP в опухоли A549 после лечения. a Анализ транскрипции TP методом ПЦР в реальном времени. Нормализовано по β-актину. b Вестерн-блот анализ экспрессии TP. в качестве внутреннего контроля использовался β-актин. Опухоли A549 резецировали у мышей BALB/c-nu через 7 дней после лечения DCA, 7,5 мг/день капецитабина, DCA плюс 7,5 мг/день капецитабина или контрольной группы. Образцы из трех опухолей каждой группы были объединены вместе
Рисунок 6. Транскрипция и экспрессия TP в опухоли A549 после лечения. a Анализ транскрипции TP методом ПЦР в реальном времени. Нормализовано по β-актину. b Вестерн-блот анализ экспрессии TP. в качестве внутреннего контроля использовался β-актин. Опухоли A549 резецировали у мышей BALB/c-nu через 7 дней после лечения DCA, 7,5 мг/день капецитабина, DCA плюс 7,5 мг/день капецитабина или контрольной группы. Образцы из трех опухолей каждой группы были объединены вместе
Обсуждение
DCA отдельно или в комбинации с другими методами лечения был протестирован в клинических испытаниях; однако нет сообщений о противоопухолевом действии DCA в комбинации с капецитабином. Мы предположили, что стимулирующее апоптоз действие DCA на клетки солидных опухолей сделает их более чувствительными к капецитабину. В настоящем исследовании только 1,4 г/л DCA не оказывал значительного влияния на рост опухоли NSCLC A549 у мышей. Однако совместное введение DCA и капецитабина позволило снизить эффективную дозу капецитабина на 50 %. DCA усиливал противоопухолевый эффект капецитабина in vivo через сенсибилизацию апоптоза. DCA оказывает незначительное влияние на метаболизм капецитабина, что не увеличивает токсичность капецитабина.
Поскольку были получены как положительные, так и отрицательные результаты (как показано в разделе «Введение»), до сих пор существуют разногласия по поводу использования DCA в качестве противоракового агента. Это расхождение может быть связано с различными экспериментальными условиями, особенно между результатами in vivo и in vitro. Клеточные анализы, использованные в настоящем исследовании, показали, что опухолевые клетки нечувствительны к DCA при культивировании in vitro, IC50 превышает 50 мМ. (данные не показаны). Действие DCA на раковые клетки не связано с прямой цитотоксичностью, а зависит от метаболического паттерна раковых клеток [7, 28]. Клеточные линии, культивируемые in vitro, не могут воспроизвести микроокружение опухоли и могут утратить «эффект Варбурга». Поэтому клеточные анализы in vitro могут быть не самыми подходящими модельными системами для определения применения DCA. Поэтому в настоящем исследовании комбинированный противоопухолевый эффект DCA и капецитабина был оценен на мышиных моделях опухолей in vivo. Мы также обнаружили, что эффект DCA на ксенотрансплантат NSCLC A549 в настоящем исследовании был меньше, чем в работе Bonnet et al. [7], даже несмотря на использование большей дозы DCA. Это может быть связано с разницей в метаболической способности ДКА у мышей и крыс. Полученные результаты свидетельствуют о том, что на эффект ДКА может влиять состояние раковых клеток и состояние пациентов, что требует тщательного изучения при клиническом использовании ДКА в лечении рака.
В настоящем исследовании на животных моделях было показано, что DCA сам по себе проявляет слабый противоопухолевый эффект против установленных опухолей. Но в сочетании с капецитабином DCA резко усиливал противоопухолевый эффект капецитабина, что видно как из исследований на животных моделях, так и из результатов иммуногистохимии апоптоза. В соответствии с этим, анализ вестерн-блотов показал, что DCA сам по себе оказывает незначительное влияние на экспрессию и расщепление прокаспаз. Но в сочетании с капецитабином DCA значительно увеличивает экспрессию и расщепление прокаспаз 8, 9 и 3. Сообщается, что цитотоксический агент, такой как 5-Fu, может привести к увеличению экспрессии и активации каспазы 8 [29,30]. Причина, по которой капецитабин приводит к увеличению экспрессии и активации каспазы 9, до сих пор не ясна. Возможно, это связано с антиангиогенетическим действием капецитабина. Сообщается, что DCA может нормализовать ось митохондрий-K+ каналов и действовать как сенсибилизатор апоптоза [7]. Мы предполагаем, что DCA может деполяризовать митохондриальный потенциал раковых клеток, нормализуя эту ось, и тем самым усиливать активацию каспазы 8 и каспазы 9 капецитабином. Повышенная активация каспазы 8 и каспазы 9 приводит к повышенной активации каспазы 3.
Недавно был подчеркнут критический вклад CSCs с их повышенной опухолеродной способностью и устойчивостью к радио- и химиотерапии в злокачественное поведение [31]. Сообщается, что CSCs в солидных опухолях играют важную роль в антихимиотерапевтических и антирадиотерапевтических характеристиках опухолей [32, 33]. Обсуждается множество методов лечения, направленных на CSCs [34, 35], а комбинированные методы лечения, направленные как на CSCs, так и на «нормальные» раковые клетки, вызывают большой интерес [36-38]. Сообщается, что DCA индуцирует апоптоз в предполагаемых стволовых клетках глиобластомы как in vitro, так и in vivo [9]. В нашем исследовании после 7 дней лечения DCA количество CD133-положительных клеток в опухолевых срезах A549, определенных с помощью иммуногистохимии, уменьшилось до 0,5 % по сравнению с 6 % в контрольной группе (данные не показаны), что позволяет предположить, что DCA может также влиять на индуцирование апоптоза в КСК рака легких. Мы предполагаем, что DCA может сенсибилизировать опухолевые клетки, особенно КСК, к капецитабину. Комбинация DCA и капецитабина действует против опухолевых клеток двумя способами: капецитабин нацелен на «нормальные» раковые клетки, щадя CSCs, а DCA нацелен на CSCs и способствует апоптозу капецитабина. Другой вероятный сценарий объяснения эффекта комбинации заключается в том, что DCA подавляет ангиогенез рака in vivo [9], при котором DCA не проявляет прямого действия на раковые клетки. В дополнение к своей классической противоопухолевой активности, капецитабин может действовать как антиангиогенетическая молекула, согласно последним исследованиям [39]. DCA может усиливать антиангиогенный эффект капецитабина, что объясняет противоопухолевый эффект комбинации. Для определения подробного механизма будут проведены углубленные исследования.
В заключение, мы использовали модели опухолей, привитых сингенетически, и ксенотрансплантированных опухолей для изучения комбинированного противоопухолевого эффекта DCA и капецитабина и обнаружили, что DCA впервые потенцировал противоопухолевый эффект капецитабина. Мы определили, что DCA обладает способностью сенсибилизировать раковые клетки и усиливать апоптотическое действие капецитабина. При комбинированном применении DCA позволял снизить эффективную дозу капецитабина без увеличения токсичности. Недорогой, непатентованный и пероральный DCA в сочетании с пероральным капецитабином может стать хорошей терапевтической схемой против рака.
Благодарности
Мы очень благодарны Венлонг Рену из Шанхайского института фармацевтической промышленности за помощь в подготовке мышиных опухолевых моделей.
Конфликт интересов
Нет.





