Амигдалин блокирует in vitro адгезию и инвазию клеток почечно-клеточной карциномы с помощью интегрин-зависимого механизма
Амигдалин блокирует in vitro адгезию и инвазию клеток
почечно-клеточной карциномы с помощью интегрин-зависимого механизма
- Автор:
- Ева Юнгель
- Масуд Афшар
- Ясмина Макаревич
Аннотация
Информация о природном соединении амигдалине, которое используется в качестве противоопухолевого средства, скудна, и поэтому его эффективность остается спорной. В этом исследовании, чтобы определить, оказывает ли амигдалин противоопухолевое действие на клетки почечно-клеточной карциномы (ПКР), было изучено его влияние на метастатическую активность ПКР. Клеточные линии ПКР, Caki-1, KTC-26 и A498, были подвергнуты воздействию амигдалина из абрикосовых косточек, и была исследована адгезия к эндотелию сосудов человека, иммобилизованному коллагену или фибронектину. Было также определено влияние амигдалина на хемотаксическую и инвазивную активность, а также влияние амигдалина на поверхностную и общую клеточную экспрессию α и β интегринов, которые участвуют в метастазировании. Мы отметили, что амигдалин вызвал значительное снижение хемотаксической активности, инвазии и адгезии к эндотелию, коллагену и фибронектину. Используя анализ FACScan, мы отметили, что амигдалин также индуцировал снижение, особенно в интегринах α5 и α6, во всех трех клеточных линиях. Функциональное блокирование α5 привело к значительному снижению адгезии KTC-26 и A498 к коллагену, а также к снижению хемотаксического поведения во всех трех клеточных линиях. Блокирование интегрина α6 значительно снизило хемотаксическую активность во всех трех клеточных линиях. Таким образом, мы предполагаем, что воздействие амигдалина на клетки почечно-клеточного рака ингибирует метастатическое распространение и связано с подавлением интегринов α5 и α6. Поэтому мы предполагаем, что амигдалин оказывает противоопухолевую активность in vitro, и это может быть связано с регуляцией интегрина.
Введение
Почечно-клеточная карцинома (ПКР) является наиболее распространенной опухолью почки. Примерно у трети пациентов есть метастазы на момент постановки диагноза, и до 30% пациентов развивают метастазы во время терапии. После метастазирования прогноз для пациентов неутешительный. Лучшее понимание молекулярных механизмов действия, лежащих в основе развития и прогрессирования ПКР, способствовало разработке таргетной терапии, тем самым улучшая прогноз для пациентов на поздних стадиях этого заболевания. Однако, несмотря на эти терапевтические достижения, прогноз для пациентов с ПКР остается неблагоприятным, 5-летняя выживаемость составляет от 5 до 12%. Неудовлетворенность традиционной терапией и желание уменьшить побочные эффекты привели многих пациентов к комплементарной и альтернативной медицине (КАМ). До 80% онкологических больных в Соединенных Штатах и более 50% онкологических больных в Европе используют КАМ наряду с традиционной терапией или вместо нее.
Информация об эффективности природных соединений скудна, и некоторые из этих соединений, такие как цианогенный дигликозид амигдалин (D-манделонитрил-β-гентиобиозид), остаются спорными. Амигдалин получают из плодовых косточек семейства розоцветных, которое включает Prunus persica (персик), Prunus armeniaca (абрикос) и Prunus amygdalus var. amara (горький миндаль). Амигдалин, в основном в Соединенных Штатах, назначают онкологическим больным с 1920-х годов. В 1950-х годах была синтезирована и запатентована внутривенная, химически иная форма амигдалина как лаэтрил. Хотя лаэтрил отличается от амигдалина, эти термины часто используются взаимозаменяемо, что затрудняет интерпретацию данных. К 1978 году около 70 000 онкологических больных в США прошли лечение амигдалином. Однако исследования амигдалина, основанные на фактических данных, остаются ограниченными. Клиническое исследование, спонсируемое Национальным институтом рака более 30 лет назад, не выявило никаких признаков регрессии опухоли ( 1 ), тогда как ретроспективный анализ 67 пациентов с опухолями, получавших амигдалин, сообщил о двух полных и четырех частичных ответах ( 2 ). Амбивалентность также отражена в отчетах о случаях: амигдалин был неэффективен в пяти случаях и эффективен в четырех. Насколько нам известно, рандомизированные клинические испытания и последующие исследования не проводились. Сторонники считают амигдалин эффективным натуральным вариантом лечения рака, тогда как противники предупреждают о токсичности из-за метаболизма цианистого водорода.
Метастазы являются основной причиной смертности, связанной с ПКР. Трансэндотелиальная миграция и подвижное распространение являются критическими этапами в распространении и прогрессировании опухоли ( 3 ), а распространение раковых клеток в отдаленные органы представляет собой основную клиническую проблему при лечении рака. В настоящем исследовании изучалось противоопухолевое действие амигдалина на адгезию и миграционные свойства клеток ПКР. Поскольку интегрины активируют ряд внутриклеточных сигнальных путей, участвующих в пролиферации, дифференцировке и подвижности клеток, был определен паттерн экспрессии рецепторов адгезии интегрина α и β между обработанными амигдалином клетками и необработанными контрольными клетками. Интегрины важны как для здоровья, так и для болезни ( 4 ) и играют ключевую роль в канцерогенезе и прогрессировании рака ( 4 ).
Настоящее исследование основано на предыдущем исследовании, посвященном влиянию амигдалина на метастатические свойства трех линий клеток рака мочевого пузыря ( 5 ). Поскольку были обнаружены некоторые различия в действии амигдалина на метастатические свойства различных линий клеток рака мочевого пузыря, возник вопрос о том, ограничиваются ли различные эффекты амигдалина определенными опухолевыми образованиями или возникают в других. Таким образом, были выбраны три линии клеток RCC, поскольку опухоли RCC являются наиболее агрессивной урологической опухолью.
Материалы и методы
Культура клеток
Клетки карциномы почки, Caki-1, KTC-26 и A498, были приобретены у LGC Promochem GmbH (Wesel, Германия). Клетки выращивали и субкультивировали в среде RPMI-1640 (Seromed, Берлин, Германия) с добавлением 10% сыворотки плода теленка (FCS), 20 мМ буфера HEPES, 100 МЕ/мл пенициллина и 100 мкг /мл стрептомицина при 37°C в увлажненном инкубаторе с 5% CO2 . Субкультуры из пассажей 5–24 были отобраны для экспериментального использования. Эндотелиальные клетки пупочной вены человека (HUVEC) были выделены из пупочных вен человека и собраны путем ферментативной обработки диспазой (1 МЕ/мл; Gibco-Invitrogen, Карлсбад, Калифорния, США). HUVEC выращивали в среде 199 (M199; Biozol, Мюнхен, Германия), дополненной 10% FCS, 10% объединенной человеческой сыворотки, 20 мкг /мл фактора роста эндотелиальных клеток (Boehringer, Мангейм, Германия), 0,1% гепарина, 100 нг/мл гентамицина и 20 мМ буфера HEPES (pH 7,4). Субкультуры из пассажей 1–5 были отобраны для экспериментального использования. Институциональный этический комитет больницы университета Гете, Франкфурт, Германия, отказался от необходимости получения согласия, поскольку HUVEC использовались анонимно для анализов in vitro и не имели связи с данными пациентов.
Лечение амигдалиномы
Амигдалин из абрикосовых косточек (Sigma-Aldrich, Тауфкирхен, Германия) был свежерастворен в среде для культивирования клеток, а затем добавлен к опухолевым клеткам в концентрации 10 мг/мл [ранее оцененной как оптимальная концентрация ( 6 )] либо на 24 часа, либо на 2 недели (лечение применялось три раза в неделю) для оценки острого и хронического лечения. Контрольные группы оставались необработанными. Во всех экспериментах сравнивались обработанные и необработанные культуры опухолевых клеток. Для изучения токсического действия амигдалина жизнеспособность клеток определялась трипановым синим (Gibco-Invitrogen).
Адгезия опухолевых клеток
Для анализа адгезии опухолевых клеток HUVEC переносили в 6-луночные мультипланшеты (Sarstedt, Nümbrecht, Германия) в полной среде HUVEC. Когда они достигали слияния, клетки Caki-1, KTC-26 и A498 отсоединяли от культуральных колб обработкой аккутазой (PAA Laboratories, Cölbe, Германия), а затем добавляли 0,5×10 6 клеток и оставляли на монослое HUVEC на 1, 2 или 4 часа. Затем неприкрепившиеся опухолевые клетки смывали с помощью подогретого (37°C) PBS (Ca 2+ и Mg 2+ ). Оставшиеся клетки фиксировали 1% глутаральдегидом. Подсчет адгезивных опухолевых клеток производился в пяти различных полях определенного размера (5×0,25 мм2 ) с использованием фазово-контрастного микроскопа (ID03, 471202-9903; Carl Zeiss Microscopy GmbH, Геттинген, Германия), после чего рассчитывалась средняя скорость клеточной адгезии.
Присоединение к иммобилизованным белкам внеклеточного матрикса
24-луночные планшеты покрывали коллагеном G (извлеченным из телячьей кожи, состоящим из 90% коллагена типа I и 10% коллагена типа III, и разведенным до 400 мкг /мл в PBS; Biochrom, Берлин, Германия) или фибронектином (извлеченным из мышей и разведенным до 100 мкг /мл в PBS; Becton-Dickinson, Гейдельберг, Германия) на ночь. Пластиковые чашки служили фоновым контролем. Планшеты промывали 1% бычьим сывороточным альбумином (БСА) в PBS для блокирования неспецифической клеточной адгезии. Затем в каждую лунку добавляли опухолевые клетки (0,1×10 6 ) и оставляли на 30 минут для инкубации. Затем не прилипшие опухолевые клетки смывали, оставшиеся прилипшие клетки фиксировали 2% глутаральдегидом и подсчитывали микроскопически. Средний показатель клеточной адгезии, определяемый соотношением адгезивных клеток, хорошо покрытых слоем, и адгезивных клеток, фон, был рассчитан по пяти различным полям наблюдения.
Хемотаксическая активность
Сывороточно-индуцированное хемотаксическое движение исследовали с использованием 6-луночных камер Transwell (Greiner, Frickenhausen, Германия) с порами 8 мкм . Клетки (0,5×10 6 клеток Caki-1, KTC-26 или A498/мл) помещали в верхнюю камеру в бессывороточной среде, либо без амигдалина (контроль), либо содержащей амигдалин. Нижняя камера содержала 10% сыворотки. После ночной инкубации верхнюю поверхность мембраны transwell осторожно протирали ватным тампоном, чтобы удалить клетки, которые не мигрировали. Клетки, перемещающиеся на нижнюю поверхность мембраны, окрашивали гематоксилином и подсчитывали микроскопически. Средняя скорость миграции рассчитывалась из пяти различных полей наблюдения.
Вторжение
Вторжение исследовали с помощью хемотаксического движения, вызванного сывороткой, через мембрану (Greiner) с порами 8 мкм , предварительно покрытую коллагеном G (извлеченным из телячьей кожи, состоящим из 90% коллагена типа I и 10% коллагена типа III; разбавленным до 400 мкг /мл в PBS; Biochrom) и HUVEC, выращенными до слияния. Клетки Caki-1, KTC-26 или A498 (0,5×10 6 /мл) помещали в верхнюю камеру в бессывороточной среде, либо без амигдалина (контроль), либо содержащей амигдалин. Нижняя камера содержала 10% сыворотки. После инкубации в течение ночи верхнюю поверхность мембраны transwell осторожно протирали ватным тампоном, чтобы удалить клетки, которые не мигрировали. Клетки, которые переместились на нижнюю поверхность мембраны, окрашивали гематоксилином и подсчитывали микроскопически. Средний показатель миграции рассчитывался в пяти различных полях наблюдения.
Экспрессия поверхности интегрина
Опухолевые клетки промывали в блокирующем растворе (PBS, 0,5% BSA), а затем инкубировали в течение 60 мин при 4°C с конъюгированными с фикоэритрином (PE) моноклональными антителами, направленными против следующих подтипов интегринов: анти-α1 (мышиный IgG1; клон SR84; #559596), анти-α2 (мышиный IgG2a; клон 12F1-H6; #555669), анти-α3 (мышиный IgG1; клон C3II.1; #556025), анти-α4 (мышиный IgG1; клон 9F10; #555503), анти-α5 (мышиный IgG1; клон IIA1; #555617), анти-α6 (мышиный IgG2a; клон GoH3; #555736), анти-β1 (мышиный IgG1; клон MAR4; #555443), анти-β3 (мышиный IgG1; клон VI-PL2; #555754) или анти-β4 (крысиный IgG2b; клон 439-9B; #555720) (все от BD Pharmingen, Гейдельберг, Германия). Экспрессия интегрина опухолевых клеток затем измерялась с помощью FACScan (BD Biosciences, Гейдельберг; анализ гистограммы канала FL-2H (log); 1×10 4 клеток/сканирование) и выражалась как средняя относительная интенсивность флуоресценции (RFI). В качестве изотипических контролей использовали мышиный IgG1-PE (MOPC-21; #555749), IgG2a-PE (G155-178; #555574) и крысиный IgG2b-PE (R35-38; #555848; все от BD Biosciences).
вестерн-блоттинг
Для исследования содержания интегрина лизаты опухолевых клеток наносили на 7–12% полиакриламидный гель (в зависимости от размера белка) и подвергали электрофорезу в течение 90 мин при 100 В. Затем белок переносили на нитроцеллюлозные мембраны. После блокирования обезжиренным сухим молоком в течение 1 часа мембраны инкубировали в течение ночи со следующими антителами: интегрин α1 (кроличий, поликлональный, 1:1000; #AB1934; Chemicon/Millipore GmbH, Швальбах, Германия), интегрин α2 (мышиный IgG1, 1:250, клон 2; #611017; BD Biosciences), интегрин α3 (кроличий, поликлональный, 1:1000; #AB1920; Chemicon/Millipore GmbH), интегрин α4 (мышиный, 1:200, клон: C-20; #sc-6589; Santa Cruz Biotechnology, Inc., Санта-Крус, Калифорния, США)], интегрин α5 (мышиный IgG2a, 1:5000, клон 1; #610634; BD Biosciences), интегрин α6 (кролик, 1:200, клон H-87; #sc-10730; Santa Cruz Biotechnology, Inc.) и интегрин β1 (мышиный IgG1, 1:2500, клон 18; #610468), интегрин β3 (мышиный IgG1, 1:2500, клон 1; #611141) и интегрин β4 (мышиный IgG1, 1:250, клон 7; #611233) (все от BD Biosciences). Конъюгированные с HRP козьи антимышиные IgG и конъюгированные с HRP козьи антикроличьи IgG (оба 1:5000; Upstate Biotechnology, Лейк-Плэсид, штат Нью-Йорк, США) служили в качестве вторичных антител. Кроме того, сигнализация, связанная с интегрином, была исследована с помощью антител к антиинтегрин-связанной киназе (ILK) (клон 3, разведение 1:1000; № 611803), антифокальной адгезионной киназе (FAK) (клон 77, разведение 1:1000; № 610088) и анти-p-специфической FAK (pY397; клон 18, разведение 1:1000; № 611807) (все от BD Biosciences). Конъюгированные с HRP козьи антимышиные IgG (разведение 1:5000; Upstate Biotechnology) служили в качестве вторичных антител. Мембраны были кратковременно инкубированы с реагентом для обнаружения ECL (ECL™; Amersham, GE Healthcare, Мюнхен, Германия) для визуализации белков, а затем проанализированы с помощью системы Fusion FX7 (Peqlab, Эрланген, Германия). β-актин (1:1000; Sigma-Aldrich) служил в качестве внутреннего контроля.
Для анализа плотности пикселей белковых полос использовалось программное обеспечение Gimp 2.8. Было рассчитано соотношение интенсивности белка/интенсивности β-актина, выраженное в процентах по отношению к контрольным значениям, принятым за 100%.
Блокировка экспериментов
Чтобы определить, влияют ли интегрины α5 и α6 на метастатическое распространение независимо от амигдалина в клеточных линиях Caki-1, KTC-26 и A498, клетки инкубировали в течение 60 мин с 10 мкг /мл мышиных моноклональных антител против интегрина α5 (клон P1D6) или крысиных моноклональных антител против интегрина α6 (клон NKI-GoH3) (оба от Millipore). Контрольные образцы инкубировали только с клеточной культуральной средой. Затем адгезию опухолевых клеток к иммобилизованному коллагену, а также хемотаксис анализировали, как описано выше.
Статистический анализ
В настоящем исследовании все эксперименты проводились 3–6 раз. Статистическая значимость определялась с помощью U-критерия Вилкоксона, Манна-Уитни. Значение p < 0,05 считалось указывающим на статистически значимое различие.
Результаты
Амигдалин блокирует взаимодействие между эндотелием опухолевых клеток и матриксом опухолевых клеток
После 24 часов обработки амигдалином адгезия опухолевых клеток A498 к HUVEC значительно снизилась, но адгезия клеток Caki-1 и KTC-26 не снизилась (по сравнению с необработанными контролями, принятыми за 100%) ( рис. 1 ). Увеличение времени воздействия амигдалина до 2 недель значительно снизило адгезию к HUVEC во всех трех линиях опухолевых клеток ( рис. 1 ).
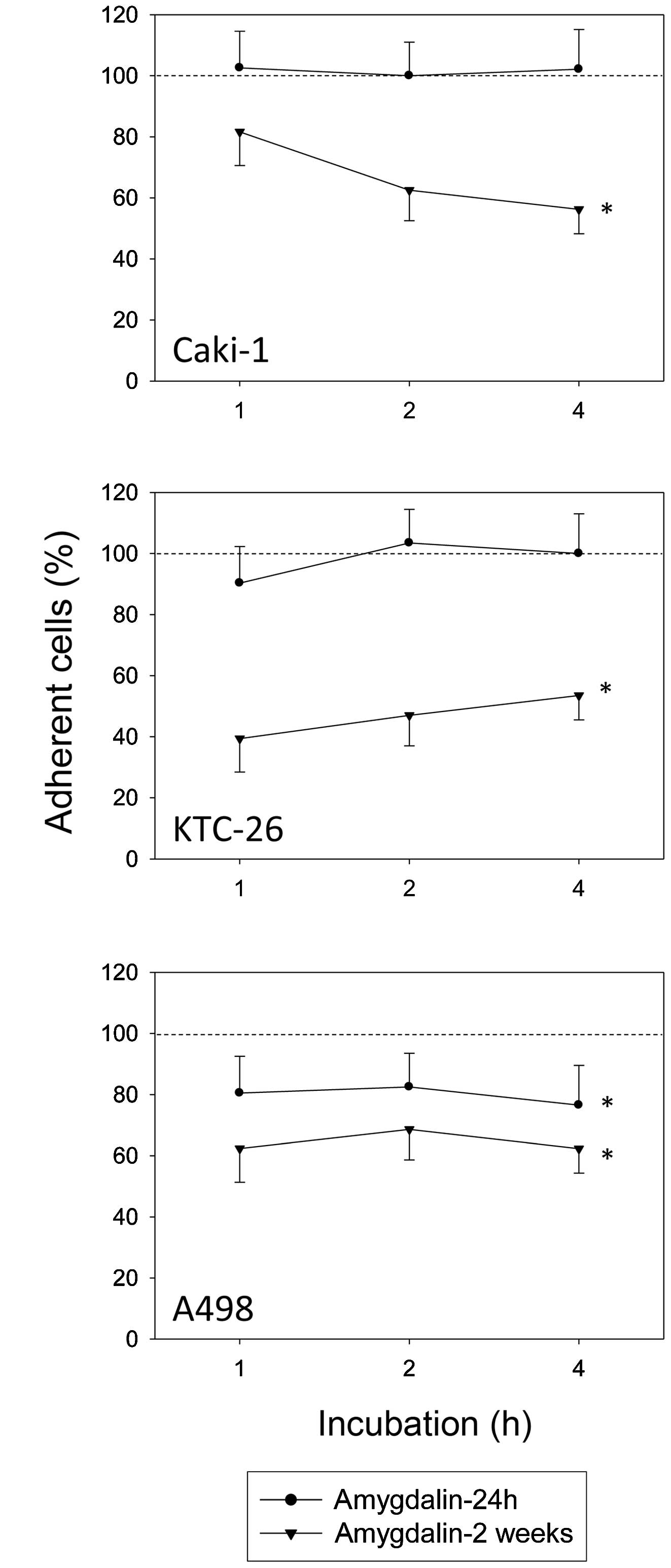 |
Рисунок 1Адгезия Caki-1, KTC-26 и A498 к эндотелию (эндотелиальные клетки пуповины человека; HUVEC). Опухолевые клетки обрабатывали 10 мг/мл амигдалина в течение 24 ч или 2 недель. Контрольные образцы оставались необработанными. Опухолевые клетки (0,5×106 клеток/лунку) добавляли и оставляли на монослое HUVEC в течение 1, 2 или 4 ч. Оценивалось среднее количество адгезированных опухолевых клеток из пяти полей, а также рассчитывался процент обработанных клеток почечно-клеточной карциномы (RCC) по сравнению с необработанными контрольными клетками (принято за 100%, пунктирная линия). *Значительное различие с контрольными образцами. Полоски указывают среднее значение ± стандартное отклонение (SD). n=5 экспериментов, p≤0,05. |
Амигдалин вызвал значительное снижение связывающей способности всех трех линий клеток RCC с иммобилизованным коллагеном и фибронектином по сравнению с контрольной группой ( рис. 2 ). Прикрепление всех трех линий клеток к матричным белкам было снижено через 24 часа, а также через 2 недели. В клеточной линии KTC-26 кратковременное применение амигдалина (24 часа) вызвало большее снижение адгезии, чем долгосрочное применение амигдалина (2 недели).
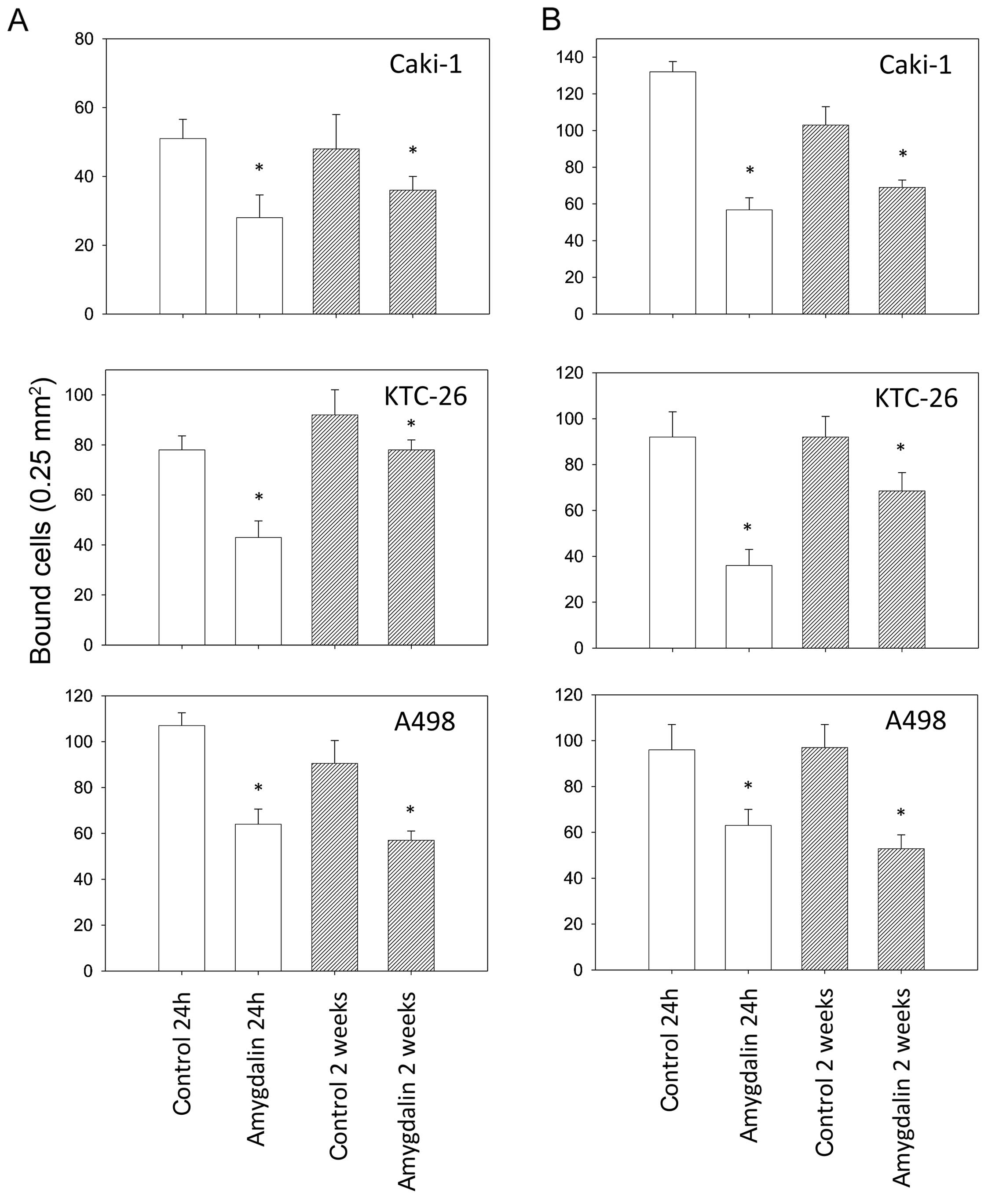 |
Рисунок 2(A) Адгезия Caki-1, KTC-26 и A498 к иммобилизованному коллагену (слева) и (B) фибронектину (справа). Опухолевые клетки обрабатывали 10 мг/мл амигдалином в течение 24 ч или 2 недель. Необработанные клетки почечно-клеточной карциномы (ПКР) служили в качестве контроля. Клетки (0,1×106 клеток/лунка) добавляли к иммобилизованному коллагену или фибронектину. Среднее количество адгезированных опухолевых клеток из пяти полей рассчитывали через 30 мин. *Значительное различие с контролем. Полоски указывают среднее значение ± стандартное отклонение (SD). n=6 экспериментов, p≤0,05. |
Амигдалин изменяет подвижность опухолевых клеток почечно-клеточного рака
Хемотаксическая активность клеток Caki-1, KTC-26 и A498 значительно снизилась после 24 ч и 2 недель применения амигдалина по сравнению с необработанными контрольными клетками ( рис. 3A ). Инвазия опухолевых клеток через покрытые коллагеном мембраны transwell также значительно снизилась в клетках Caki-1 и A498 после 24 ч и 2 недель применения амигдалина ( рис. 3B ). Однако 2 недели применения амигдалина не снизили инвазивную способность клеток KTC-26.
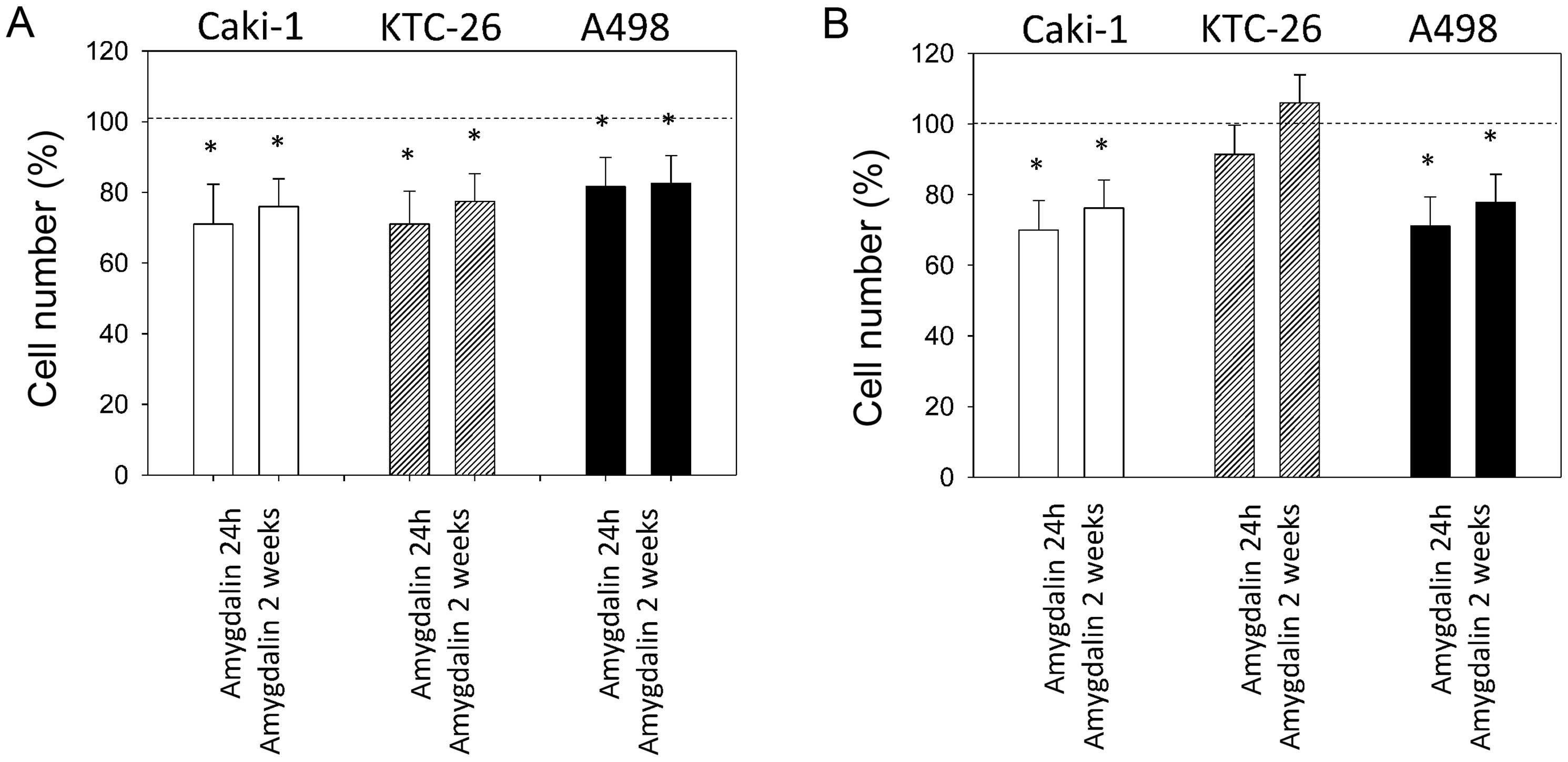 |
Рисунок 3Влияние амигдалина на клетки почечно-клеточной карциномы (ПКР): хемотаксис (A) и инвазия (B). Клетки ПКР, обработанные амигдалином в течение 24 ч или 2 недель, высевали в верхнюю камеру с хемоаттрактантом в лунке ниже. Подсчитывали клетки, мигрирующие через мембрану через 20 ч. Контрольные клетки не получали амигдалин и были установлены на 100% (пунктирная линия). *Значительное различие с контрольными клетками. Полоски указывают среднее значение ± стандартное отклонение (SD). n=5 экспериментов, p≤0,05. |
Амигдалин модулирует поверхностную экспрессию интегрина α и β
Клетки Caki-1, KTC-26 и A498 характеризовались различными базальными поверхностными паттернами экспрессии интегринов α и β ( рис. 4A ). Caki-1 заметно экспрессировал α3 и β1, умеренно экспрессировал α5 и β3, тогда как α1, α2, α4, α6 и β4 были лишь незначительно обнаружены. KTC-26 сильно экспрессировал α3 и β1. Члены подтипа α1, α2, α5, α6, β3 и β4 были умеренно экспрессированы, а α4 не был обнаружен. Профиль экспрессии интегринов A498 был похож на профиль KTC-26, за исключением α4, который был обнаружен для A498. Мы также отметили, что β4 присутствовал в клетках KTC-26, но не в клетках A498.
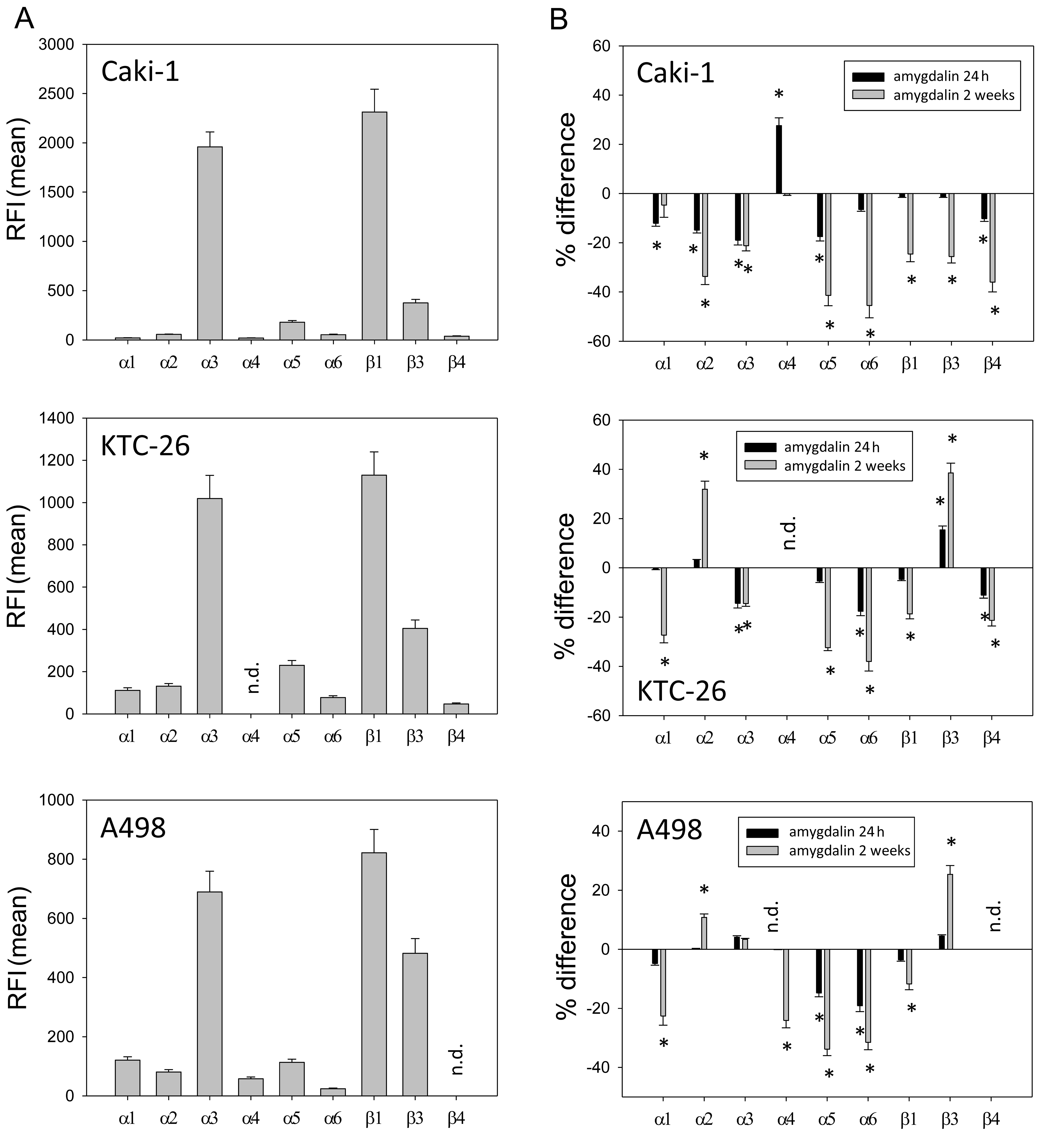 |
Рисунок 4(A) Базальная поверхностная экспрессия подтипов интегрина почечно-клеточной карциномы (RCC) и (B) разница (%) с необработанным контролем через 24 часа или 2 недели воздействия амигдалина. RFI, относительная интенсивность флуоресценции. nd, не обнаруживается. *Значительная разница с контролем. Полоски указывают среднее значение ± стандартное отклонение (SD), p≤0,05. |
Применение амигдалина в течение двадцати четырех часов и в течение двух недель изменило профиль поверхности интегрина, который является специфическим для типа клеток ( рис. 4B ). Мы отметили, что α5 и α6 были значительно подавлены во всех клеточных линиях после двух недель воздействия амигдалина. β1, который был сильно выражен во всех трех клеточных линиях, также был значительно снижен после двух недель применения амигдалина. Высокая базальная экспрессия рецептора α3 была снижена в Caki-1 и KTC-26, но не в A498, под действием амигдалина. Различия также были отмечены в отношении α2 и β3, оба из которых были снижены в клетках Caki-1, но повышены в клетках KTC-26 и A498 через две недели. Сниженные уровни экспрессии β4 были обнаружены в клетках Caki-1 и KTC-26 после воздействия амигдалина.
Амигдалин влияет на общее содержание клеточных интегринов
Оценка содержания белка интегрина после 24 ч воздействия амигдалина выявила значительную положительную регуляцию α2 и отрицательную регуляцию α3 и p-FAK ( рис. 5 ). β1 был значительно повышен в Caki-1 и KTC-26, тогда как α6 был значительно снижен в Caki-1 и A498, а общее содержание α5 было снижено в клетках A498 через 24 ч. В клетках Caki-1 α4 и β4 увеличились, а β3 уменьшился.
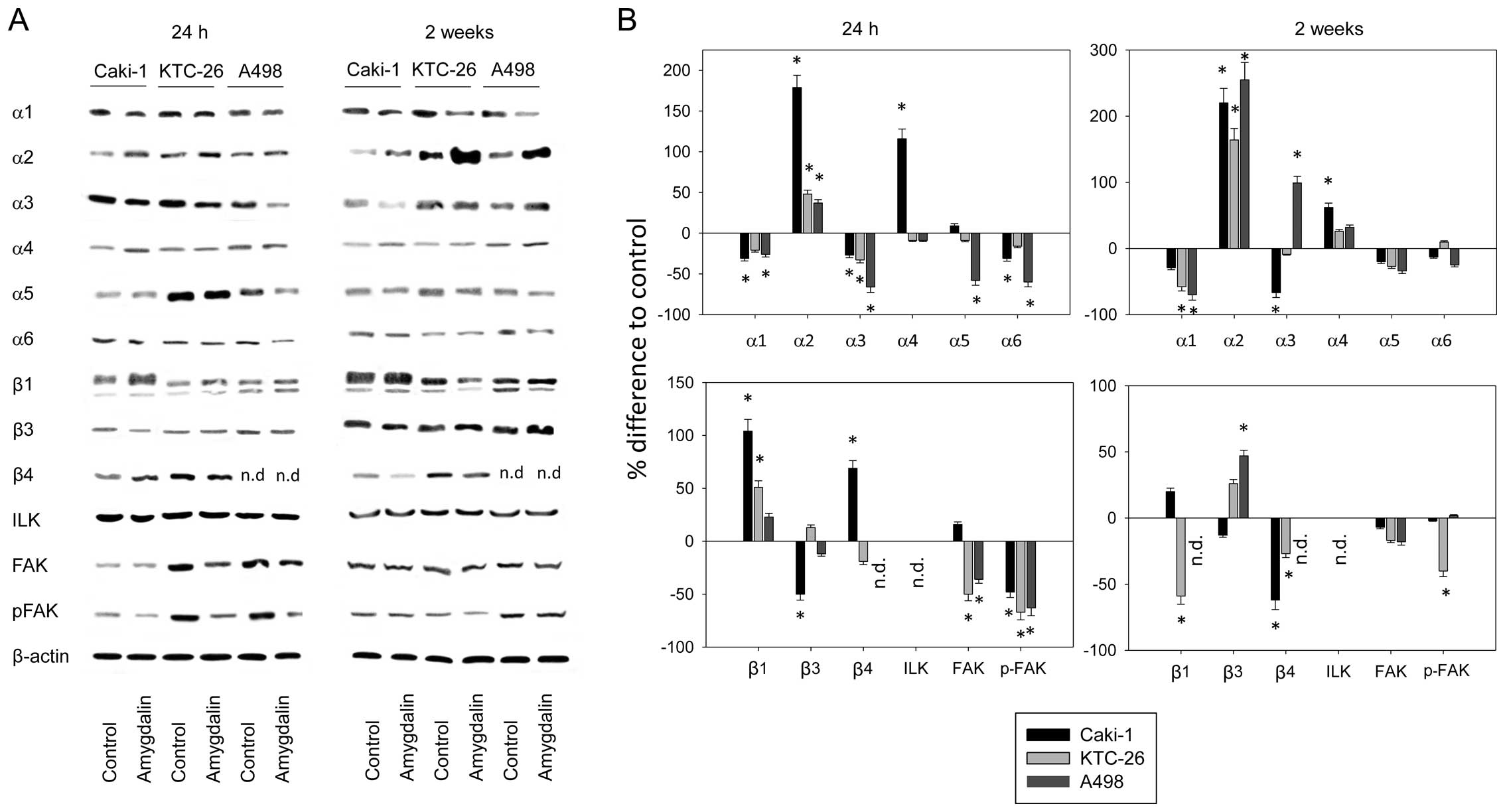 |
Рисунок 5(A) Вестерн-блот-анализ общего содержания интегрина в клетках Caki-1, KTC-26 и A498, подвергнутых воздействию амигдалина в течение 24 ч или 2 недель, и необработанных контролях. В качестве внутреннего контроля использовался β-актин. (B) Анализ плотности пикселей белковых полос вестерн-блоттинга. Соотношение интенсивности белка/интенсивности β-актина было рассчитано и выражено в процентах по отношению к контролям, установленным на 100% (0) после того, как клетки подвергались воздействию амигдалина в течение 24 ч или 2 недель. Показан один представитель из трех отдельных экспериментов. nd, не обнаружено. *Значительное различие с контролями, p≤0,05. |
После 2 недель воздействия амигдалина интегрин α2 значительно увеличился, даже больше, чем после воздействия в течение 24 часов. В клетках A498 α3 и β3 увеличились после 2 недель воздействия амигдалина. Снижение β1 и p-FAK произошло в клетках KTC-26, а β4 был подавлен как в Caki-1, так и в KTC-26 после 2 недель воздействия амигдалина ( рис. 5 ).
Блокировка экспериментов
Профили экспрессии интегрина всех трех линий клеток были изменены амигдалином. Поскольку поверхностный интегрин α5 и интегрин α6 были сильно снижены во всех трех линиях клеток после применения амигдалина, эти интегрины были выбраны для исследований функциональной блокировки, чтобы выяснить, коррелируют ли эти снижения с изменениями адгезии и миграции опухолевых клеток. Блокирование α5 привело к значительному ингибированию адгезии клеток KTC-26 и A498 к коллагену ( рис. 6A ). Однако адгезия клеток Caki-1 к коллагену не была существенно затронута. Блокирование интегрина α5 привело к снижению хемотаксической активности во всех трех линиях клеток ( рис. 6B ). Блокирование рецептора α6 не оказало существенного влияния на адгезию к коллагену ни в одной из линий клеток ( рис. 6A ), но значительно снизило хемотаксис во всех трех линиях клеток ( рис. 6B ).
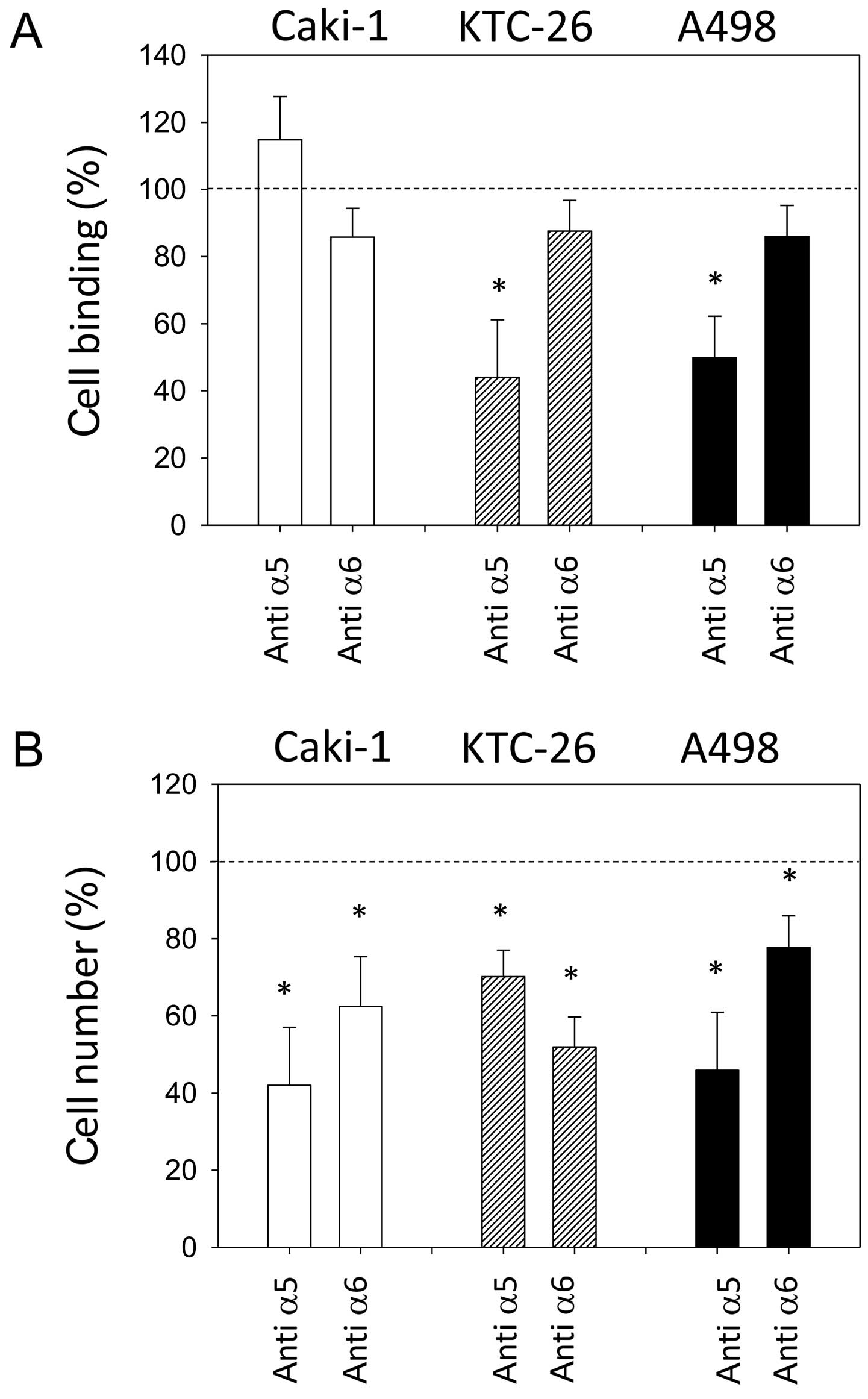 |
Рисунок 6Влияние функциональной блокировки интегрина α5 и α6 на (A) адгезию клеток к коллагену и (B) хемотаксис. Неблокированные клетки служили в качестве контроля (100%, пунктирная линия). Оценивалось среднее количество адгезированных или хемотаксически активных клеток из пяти полей (0,25 мм2). Полосы указывают среднее значение ± стандартное отклонение (SD). *Значительное различие с контролем. n=3 эксперимента, p≤0,05. |
Обсуждение
Было отмечено, что взаимодействие между опухолевыми клетками и эндотелием играет решающую роль в метастатической прогрессии; адгезия клеток немелкоклеточного рака легких (НМРЛ) к эндотелию стенки сосудов была связана с трансмиграцией опухолевых клеток, что приводит к метастазам в мозг и лимфатические узлы ( 7 ). Более агрессивный, метастазирующий фенотип рака также был связан с повышенной адгезией ( 7 ). В настоящем исследовании мы продемонстрировали, что воздействие амигдалина привело к значительному ингибированию связывающего взаимодействия между клетками RCC и монослоем HUVEC, коллагеном и фибронектином. Таким образом, была ингибирована хемотаксическая и инвазивная активность клеток RCC. Такое ингибирование является клинически значимым, поскольку трансэндотелиальная миграция и подвижное распространение являются критическими этапами в распространении и прогрессировании опухоли ( 3 ) и коррелируют с плохой выживаемостью. Снижение миграционного потенциала было связано с успешной терапией опухолей и менее злокачественным фенотипом опухоли ( 8 ). Таким образом, мы предполагаем, что ингибирование адгезии и подвижности клеток почечно-клеточного рака амигдалином снижает распространение метастазов.
Эффект амигдалина на блокирование адгезии и миграции не ограничивается клетками почечно-клеточного рака. Недавно было показано, что амигдалин также подавляет адгезивное поведение клеток рака мочевого пузыря ( 5 ). Хотя амигдалин оказывал схожее подавляющее действие на адгезионные свойства клеток рака мочевого пузыря и почечно-клеточного рака, миграция клеток рака мочевого пузыря была затронута амигдалином по-разному. Хемотаксис был подавлен в двух, но повышен в одной линии клеток рака мочевого пузыря после воздействия амигдалина, что указывает на то, что влияние амигдалина, вероятно, зависит от сущности опухоли. Таким образом, важно исследовать влияние амигдалина на различные сущности опухоли.
Семейство интегринов вовлечено во все этапы метастатической прогрессии опухоли ( 4 , 7 ). Интегрин α5 повышается в опухолевых клетках эпителиального происхождения, и была установлена положительная корреляция между экспрессией интегрина α5 и адгезией клеток почечно-клеточного рака ( 9 ). В настоящем исследовании введение амигдалина значительно снизило поверхностный интегрин α5 во всех трех клеточных линиях. Кроме того, общее клеточное содержание интегрина α5 в зависимости от времени снижалось в присутствии амигдалина. Блокирование функции интегрина α5 вызвало значительное ингибирование адгезии клеток KTC-26 и A498 к коллагену и снижение хемотаксической активности всех используемых клеточных линий. В соответствии с настоящими данными, снижение регуляции интегрина α5 ранее было связано со снижением адгезивного и инвазивного поведения нескольких типов раковых клеток ( 10–12 ) .
В настоящем исследовании мы отметили, что адгезия коллагена клеток Caki-1 не снизилась после блокирования интегрина α5, в отличие от вызванного амигдалином снижения в клетках A498 и KTC-26. Подобное различие в функции интегрина между типами опухолевых клеток наблюдалось ранее. Было показано, что блокирование интегрина α5 ингибирует взаимодействие клеток с матриксом клеток рака мочевого пузыря HCV29 и BC3726, но усиливает связывание линий клеток рака мочевого пузыря T24 и Hu456 (оснащенных другим набором интегринов) ( 13 ). Аналогичным образом, было показано, что блокирование интегрина β1 ингибирует прикрепление клеток рака мочевого пузыря UMUC-3 к коллагену, но оказывает противоположный адгезивный эффект на клетки TCCSUP, которые характеризуются другим профилем экспрессии интегринов ( 5 ). В настоящем исследовании воздействие амигдалина вызвало увеличение поверхностного β3 интегрина в клетках KTC-26 и A498, но уменьшение в клетках Caki-1. Контррегуляция с участием другого подтипа интегрина, в данном случае β3, может объяснить, почему блокирование α5 не остановило адгезию в клетках Caki-1. Потеря интегрина α5 вызвала снижение хемотаксиса во всех трех клеточных линиях. Поэтому мы предполагаем, что потеря поверхностного интегрина α5 является механизмом, посредством которого амигдалин действует на миграцию клеток RCC, а тонкая настройка производительности интегрина зависит от конкретного профиля интегрина в конкретной клеточной линии.
Было показано, что интегрин α6 облегчает миграцию эпителиальных клеток и коррелирует с риском прогрессирования, метастазирования и смерти в клинических испытаниях ( 14 ). Другие исследования показали, что интегрин α6 способствует миграции и инвазии при колоректальном раке ( 15 ), а также карциномах поджелудочной железы ( 16 ) и молочной железы ( 17 ). Было отмечено, что интегрин α6 активирует FAK ( 18 ) и связанную с FAK нисходящую сигнализацию, которая имеет отношение к контролю подвижности клеток, выживаемости и пролиферации ( 4 ). В настоящем исследовании поверхностная экспрессия интегрина α6 была значительно снижена, а FAK был дезактивирован амигдалином во всех трех клеточных линиях. Блокирование поверхностного интегрина α6 показало, что α6 не мешает адгезии опухолевых клеток, но регулирует подвижность клеток. Поэтому вполне вероятно, что снижение α6 представляет собой механизм, с помощью которого амигдалин замедляет распространение клеток.
Было показано, что интегрин β1 способствует инвазии клеток при раке молочной железы, легких, поджелудочной железы и колоректальном раке, а также при глиоме, меланоме ( 4 ) и нейробластоме ( 19 ). В клетках рака предстательной железы было показано, что повышение уровня интегрина β1 сопровождается повышенным подвижным поведением, тогда как блокада интегрина β1 способствует снижению хемотаксиса, миграции и адгезии клеток ( 10 ). Ингибирование интегрина β1 было связано с уменьшением инвазии и метастазирования рака яичников ( 12 ), а ингибитор пептида интегрина α5β1, как было показано, блокирует метастазирование рака молочной железы in vivo ( 20 ). В настоящем исследовании поверхностный интегрин β1 был снижен во всех трех линиях опухолевых клеток после 2 недель воздействия амигдалина. Таким образом, амигдалин может воздействовать на интегрин β1, замедляя подвижное распространение клеток почечно-клеточного рака.
Исследования in vitro и in vivo показывают, что α3 является еще одним интегрином, участвующим в инвазии глиомы, меланомы, гепатоцеллюлярной и молочной карциномы, а также способствует метастазированию клеток молочной железы в легкие ( 4 ). Блокирование интегрина α3 привело к ингибированию адгезии клеток рака простаты ( 21 ). В настоящем исследовании поверхностный интегрин α3 был снижен в клетках Caki-1 и KTC-26, но не в клетках A498. Это неоднородное снижение указывает на специфичный для клеточной линии эффект, вызванный амигдалином.
Применение амигдалина, помимо модуляции экспрессии поверхностного интегрина, также изменило общее содержание клеточного интегрина. В настоящем исследовании общая экспрессия клеточного интегрина α2 была повышена во всех трех линиях опухолевых клеток после воздействия амидалина. Предыдущие исследования показали, что снижение уровня интегрина α2 в опухолевых клетках потенциально увеличивает распространение опухолевых клеток, а повторная экспрессия интегрина α2, как было показано, обращает вспять злокачественные свойства клеток рака молочной железы ( 22 ). Следовательно, мы предполагаем, что индуцированное амигдалином ингибирование адгезии и миграции опухолевых клеток, наблюдаемое здесь, связано с повышением регуляции интегрина α2.
Общий клеточный интегрин α3 снизился через 24 ч после нанесения амигдалина во всех трех клеточных линиях. Это согласуется с вызванным амигдалином снижением поверхностного α3 в клетках Caki-1 и KTC-26. Поскольку поверхностный α3 не был изменен в клетках A498, возможно, что амигдалин в этой клеточной линии действует через внутриклеточные сигнальные пути α3. p-FAK, который был сильно снижен в клетках A498 через 24 ч после нанесения амигдалина, подтверждает эту гипотезу, поскольку ось α3-FAK участвует в инициации и прогрессировании рака ( 23 ). Ли и др. продемонстрировали, что FAK является критическим медиатором опухолеобразования и метастазирования, которые частично зависят от интегрина α3 ( 24 ). Поэтому мы предполагаем, что снижение как интегрина α3, так и FAK дезактивирует двигательный аппарат клеток A498.
Общий клеточный интегрин β1 был повышен в клетках Caki-1 после применения амигдалина, в то время как поверхностная экспрессия была снижена. Этот тип сдвига не является необычным и указывает на транслокацию рецептора с поверхности во внутриклеточный компартмент. Хотя значимость этого процесса не полностью понята, было показано, что перемещение интегрина β1 к плазматической мембране увеличивает метастатический потенциал клеток RCC, тогда как остановка рециркуляции β1 путем поддержания высокого содержания цитоплазматической и низкого содержания плазматической мембраны снижает метастазирование ( 25 ). Было показано, что повышение внутриклеточного β1 связано с дезактивацией FAK ( 25 ), что коррелирует с настоящими результатами.
Эффекты амигдалина на экспрессию подтипа интегрина зависели от клеточной линии, от того, было ли время применения острым (24 ч) или хроническим (2 недели) и от того, находился ли интегрин на поверхности клетки или в цитоплазме. Таким образом, молекулярный способ действия амигдалина в отношении экспрессии подтипа интегрина не является однородным и может влиять как на механическое межклеточное сцепление, запускаемое интегрином, так и на активацию биохимического пути, контролируемого интегрином.
В трех исследованных линиях клеток RCC применение амигдалина значительно снизило инвазивное и подвижное поведение. Однако 2 недели применения амигдалина не снизили инвазивную способность клеток KTC-26. Снижение было преимущественно связано с уменьшением поверхностных интегринов α5 и α6. Однако этот механизм не следует обобщать. Хотя было показано, что амигдалин также ингибирует адгезию и миграцию в клетках рака мочевого пузыря, интегрины изменяются по-разному. Интегрины α5 и α6, по-видимому, являются важной мишенью амигдалина в клетках RCC, тогда как модуляция интегринов β1 или β4 была наиболее очевидна в клетках рака мочевого пузыря ( 5 ). Были начаты дальнейшие исследования in vitro , чтобы оценить, влияет ли амигдалин также на рост клеток RCC, как это наблюдалось для клеток рака мочевого пузыря ( 6 ).
В заключение, воздействие амигдалина на клетки RCC подавляет метастатическое распространение и связано с подавлением α5 и α6 интегринов. Таким образом, амигдалин проявляет противоопухолевую активность in vitro в RCC. Эта активность in vitro должна быть оценена на модели животных.
Благодарности
Настоящее исследование было поддержано «Фондом Бригитты и Норберта Мут» и «Друзья и сторонники Франкфуртского университета Гете».