Амигдалин как химиопротекторное средство при совместном лечении с цисплатином
Амигдалин как химиопротекторное средство при совместном лечении с цисплатином
оригинал статьи: https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2022.1013692/full
ОРИГИНАЛЬНАЯ ИССЛЕДОВАТЕЛЬСКАЯ статья
Фронт. Фармакол. , 20 сентября 2022 г.Секция Экспериментальная фармакология и открытие лекарствТом 13 - 2022 | https://doi.org/10.3389/fphar.2022.1013692








- 1 Медицинский факультет Европейского университета Кипра, Никосия, Кипр
- 2 Лаборатория иммунологии опухолей и биомаркеров, Центр фундаментальных и трансляционных исследований рака, Факультет наук о жизни, Европейский университет Кипра, Никосия, Кипр
- 3 Лаборатория микроокружения опухоли, метастазирования и экспериментальной терапии, Центр фундаментальных и трансляционных исследований рака, Факультет наук о жизни, Европейский университет Кипра, Никосия, Кипр
Амигдалин — это природный гликозид, используемый в традиционной китайской медицине, который, как известно, обладает противораковыми свойствами. Несмотря на то, что противораковые свойства амигдалина хорошо известны, его воздействие на нормальные клетки не было тщательно изучено. Целью настоящего исследования было изучение возможной химиопротекторной роли амигдалина против цитотоксических эффектов химиотерапии для нормальных клеток человека. В частности, он был протестирован в сочетании с сильным химиотерапевтическим препаратом цисплатином. Человеческая неканцерогенная эпителиальная клеточная линия MCF12F, клетки человеческих фибробластов, клетки человеческого рака молочной железы MCF7 и MDA-MB-231 были обработаны цисплатином в зависимости от дозы и времени в отсутствие или в присутствии амигдалина. Когда клетки MCF12F и фибробласты подвергались предварительной обработке амигдалином с последующей обработкой цисплатином (24 ч амигдалин + 24 ч цисплатин), жизнеспособность клеток увеличивалась (22%, p < 0,001), как показано с помощью анализа МТТ. Как подтверждается проточной цитометрией, комбинированное лечение было связано с уменьшением процента поздних апоптотических клеток по сравнению с монотерапией (кратность изменения снижения = 1,6 и 4,5 для 15 и 20 мкМ соответственно). Кроме того, экспрессия белков PUMA, p53, фосфо-p53 и Bax снижалась, когда использовалась комбинированная обработка по сравнению с одним цисплатином, в то время как проапоптотические белки Bcl-2 и Bcl-xL демонстрировали повышенную тенденцию в присутствии амигдалина. Более того, уровни проапоптотических генов PUMA , p53 и мРНК BAX были значительно снижены (∼83%, ∼66% и ∼44% соответственно) по сравнению с одним цисплатином, в то время как уровни мРНК антиапоптотических генов BCl-2 и Bcl-XL были повышены (∼44,5% и ∼51% соответственно) по сравнению с одним цисплатином после 24 ч комбинированного лечения. Исследование анализа индекса комбинации (CI) показало, что амигдалин, возможно, можно рассматривать как антагонист цисплатина (2,2 и 2,3) для клеток MCF12F и фибробластов соответственно. Напротив, для клеток рака молочной железы MCF7 и MDA-MB-231 амигдалин и цисплатин показали синергический эффект (0,8 и 0,65) соответственно. Наши текущие результаты показывают, что амигдалин оказывает химиомодулирующее действие при совместном лечении с цисплатином и способен защищать нормальные клетки молочной железы, а также фибробласты во время химиотерапии, что указывает на сильную селективную химиопротекторную способность и может способствовать улучшению качества жизни онкологических больных.
Введение
Рак является второй по значимости причиной смерти в мире, после сердечно-сосудистых заболеваний ( Nagai and Kim, 2017 ). Рак молочной железы (РМЖ) является наиболее распространенным видом рака среди женщин во всем мире и представляет собой серьезную проблему для общественного здравоохранения [Всемирная организация здравоохранения (ВОЗ), 2021]. Химиотерапия является наиболее эффективным и часто используемым методом лечения большинства злокачественных новообразований ( Huang et al., 2017 ). Несмотря на многочисленные достижения за последнее десятилетие в области адъювантной терапии РМЖ, остаются нерешенными многочисленные проблемы, включая неблагоприятные побочные эффекты, вызываемые химиотерапевтическими препаратами, такие как тошнота, выпадение волос, рвота, усталость и в тяжелых случаях даже смерть ( Waris and Ahsan, 2006 ; Ye et al., 2017 ). Управление по контролю за продуктами и лекарствами США одобрило в общей сложности 132 химиотерапевтических препарата, из которых 56, как сообщается, вызывают окислительный стресс ( Chen et al., 2018 ). Многие классы химиотерапевтических препаратов, такие как таксаны и производные платины, могут вызывать окислительный стресс ( Cauli, 2021 ).
Цис-диаммининдихлорплатина (II) (цисплатин) — неорганическое соединение, «алкилирующий агент», используемый в качестве основного лечебного препарата, способного уменьшать рост раковых клеток, против различных видов рака человека, включая рак молочной железы, яичек, яичников и легких ( Dasari and Tchounwou, 2014 ). Цисплатин образует внутри- и межцепочечные аддукты с ДНК и, таким образом, является мощным индуктором остановки клеточного цикла, приводящей к апоптозу для большинства типов раковых клеток. Сшивающие взаимодействия цисплатина с ДНК способствуют ингибированию репликации, транскрипции и других ядерных функций, которые могут останавливать пролиферацию раковых клеток и рост опухоли ( Zwelling et al., 1979 ; Dasari and Tchounwou, 2014 ). Эффективность цисплатина зависит от способности клеток либо восстанавливать повреждения ДНК, либо приступать к гибели ( Mirmalek et al., 2016 ). Таким образом, сигнальные пути, регулирующие апоптоз, играют ключевую роль в том, как клетки будут реагировать на цисплатин ( Истман, 1990 ).
Амигдалин (D-манделонитрил-β-гентиобиозид) — это цианогенный диглюкозид, который естественным образом содержится в косточках многочисленных фруктов и растений семейства розоцветных, таких как Prunus armeniaca (абрикос) и Prunus persica (персик). Все больше доказательств подтверждают, что амигдалин (также известный как «лаэтрил») может действовать как противораковый агент, вызывая остановку клеточного цикла и апоптоз ( Guo et al., 2013 ; Makarevic et al., 2014 ; Lee and Moon, 2016 ; Saleem et al., 2018 ). Амигдалин проявляет синергический эффект в сочетании с другими соединениями, такими как синильная кислота (противоопухолевое вещество) и бензальдегид (обезболивающее соединение); вызывая гибель раковых клеток ( Song and Xu, 2014 ). Кроме того, in vitro сообщалось, что амигдалин связан с противораковой активностью в отношении клеток рака молочной железы, главным образом, посредством окислительного стресса; он способствует дифференциальному ингибированию пролиферации клеток MCF7 и T470 ( Abboud et al., 2019 ).
Амигдалин может быть расщеплен ферментом, известным как β -глюкозидаза, высвобождающим цианистый водород, бензальдегид и глюкозу. Бензальдегид является обезболивающим, который может быть преобразован в бензойную кислоту кислородом в нормальных/здоровых тканях. Цианистый водород может вызывать токсичность цианида и, следовательно, убивать раковые клетки ( Blaheta et al., 2016 ). С другой стороны, другой фермент, роданеза, который присутствует только в нормальных тканях, а не в раковых, по-видимому, обладает способностью детоксифицировать цианид и, следовательно, защищать нормальные ткани ( Newmark et al., 1981 ). Эти два вышеупомянутых фермента, вероятно, могут способствовать избирательной токсичности амигдалина, контролируя рост и метастазирование раковых клеток.
Теперь мы намерены изучить влияние амигдалина, цисплатина и их комбинированной терапии на жизнеспособность клеток с использованием как нормальных, так и раковых клеточных линий, а также оценить потенциальную химиопротекторную роль амигдалина.
Материалы и методы
Культура клеток
Амигдалин был приобретен у Sigma-Aldrich (Сент-Луис, Миссури, США) и растворен в воде (концентрат 1М). Концентрат цисплатина 1 мг/мл для приготовления раствора для инфузий был приобретен у Accord. Клеточные линии MCF12F, MCF7 и MDA-MB-231 были приобретены у American Type Culture Collection (ATCC, Роквилл, Мэриленд). Фибробласты, извлеченные из ткани поджелудочной железы, были любезно предоставлены Университетом Кипра. Клетки MCF12F и фибробласты культивировались в модифицированной среде Дульбекко с питательной смесью Ham's F-12 (DMEM/F12), содержащей 5% лошадиной сыворотки, обработанной Chelex, приобретенной Sigma-Aldrich, эпидермальный фактор роста (EGF, 10 мкг/500 мл), холерный токсин (50 мкг/500 мл), инсулин (5 мг/500 мл) и гидрокортизон (250 мкг/500 мл) вместе с 1% антибиотиков и антимикотиков. Клетки MCF7 и MDA-MB-231 культивировались в среде DMEM с высоким содержанием глюкозы, содержащей 10% фетальной бычьей сыворотки и 1% антибиотика, приобретенного Sigma-Aldrich. Клетки инкубировались при 37°C в увлажненной камере при 95% O2 / 5% CO2 . Bcl-2, фосфо-p53, p53, Bax, GAPDH, каспаза-9, антитела были приобретены в Cell Signalling Technology (Дэнверс, Массачусетс, США). β -Актин и каспаза-8 были приобретены в Santa Cruz Biotechnology Inc. Реагенты для культивирования клеток (DMEM, FBS, HS, антибиотик/противогрибковый препарат и трипсин) были приобретены в Gibco, Invitrogen (Карлсбад, Калифорния, США).
Анализ комбинированного индекса
Анализ индекса комбинации (CI) является самым простым способом оценки фармакологических взаимодействий препаратов и в нашем случае использовался для количественной оценки синергизма или антагонизма. Синергизм используется для описания улучшения ответа опухоли, тогда как антагонизм используется, когда эффект комбинации менее токсичен, чем результат индивидуальных эффектов.
Результаты анализа индекса комбинации основаны на теории Chou-Talalay ( Chou, 2010 ). CompuSyn — это компьютерная программа для количественной оценки синергизма и антагонизма в комбинациях лекарственных средств и определения значений IC50 и ED50. Когда этот конкретный анализ дает CI = 1, это означает, что реагирующие вещества имеют аддитивный эффект, CI < 1 означает, что реагирующие вещества имеют синергический эффект, а CI > 1 означает, что реагирующие вещества имеют антагонистический эффект ( таблица 1 и дополнительный материал 2 ) ( Chou, 2010 ).
ТАБЛИЦА 1
ТАБЛИЦА 1. Значения IC50 различных линий клеток.
МТТ-тест
Для оценки жизнеспособности клеток был проведен анализ пролиферации клеток с использованием МТТ [3-(4,5-диметилтиазол-2-ил)-2,5-дифенил тетразолия бромида, M2128 от Sigma-Aldrich] ( Gasparini et al., 2017 ). После трипсинизации и подсчета с помощью гемоцитометра клетки MCF12F и MCF7 были высеяны в 96-луночный планшет ( Green and Sambrook, 2019 ). После проверки адгезии (примерно через 18 ч после высевания) клетки инкубировали с 10 мМ амигдалина и через 24 ч добавляли 15 мкМ цисплатина еще на 24 ч. После обработки клеток 20 мкл красителя МТТ в течение 4 ч, а затем инкубации с 150 мкл ДМСО (диметилсульфоксид, D8418 от Sigma-Aldrich) в течение 15 мин. Поглощение при 595 нм измеряли с помощью микропланшетного ридера (ThermoFisher Scientific).
синтез кДНК
Общая РНК из каждой популяции клеток была выделена с помощью набора RNeasy Micro Kit (50), а концентрация, а также чистота были измерены с помощью поглощения в λ = 280 и 260 нм/280 нм соответственно. кДНК каждого образца синтезирована с помощью набора primeScript first strand cDNA Synthesis kit (Takara) для смеси 1 и 2. Смесь 1 инкубировали при 65°C в течение 10 мин, а затем во льду в течение 3 мин перед добавлением смеси 2 до конечного объема 20 мкл. Последним шагом была инкубация смесей при определенных температурах в машине RT-qPCR (Bio-Rad) ( Neophytou et al., 2019 ).
ОТ-ПЦР в реальном времени
Для измерения экспрессии мРНК была проведена ПЦР в реальном времени (RT-qPCR) с использованием набора KAPA SYBR FAST qPCR (KK4610). Были использованы следующие праймеры: Bax, прямой: 5′-ACATGGAGCTGCAGAGGATG-3′, обратный: 5′-CCAGTTGAAGTTGCCGTCAG-3′; p53, прямой: 5′-CCTCAGCATCTTATCCGAGTGG-3′, обратный: 5′-TGGATGGTGGTACAGTCAGAGC-3′; PUMA, прямой: 5′-ACGACCTCAACGCACAGTACGA-3′, обратный: 5′-GTAAGGGCAGGAGTCCCATGAT-3′; Bcl-2, прямой: 5′-GGATAACGGAGGCTGGGATG-3′, обратный: 5′-GGCCAAACTGAGCAGAGTCT-3′; Bcl-xL, прямой: 5′-AGAGCCTTGGATCCAGGAGA-3′, обратный: 5′-TCAGGAACCAGCGGTTGAAG-3′; GADPH, прямой: 5′-GTCTCCTCTGACTTCAACAGCG-3′, обратный: 5′-ACCACCCTGTTGCTGTAGCCAA-3′. GADPH использовали в качестве гена домашнего хозяйства. В конце реакции регистрировали значения Ct, и среднее значение трехкратных повторов использовали для расчета кратности изменения экспрессии гена с использованием метода 2[-Delta Delta C(T)] ( Livak and Schmittgen, 2001 ), как описано ранее ( Papageorgis et al., 2010 ). Для оценки эффекта лечения использовали данные по крайней мере трех независимых биологических повторов.
Извлечение белка
Среду удаляли из 6-луночного планшета и добавляли 1 мл PBS для промывки лунок, а затем аспирировали. После этого добавляли 150 мкл RIPA для лизиса клеток. Гомогенизированные образцы переносили в пробирки Eppendorf на льду на 30 мин с частым встряхиванием. Образцы центрифугировали при 10 000 об/мин/10 мин/4°C ( Ngoka, 2008 ). Образцы хранили при температуре –80 до дальнейшего использования ( Дополнительные материалы 1 и 3).
вестерн-блоттинг
Для определения уровня белка мы провели анализ вестерн-блоттинга. Смеси белков инкубировали при 98°C в течение 5 минут, а затем переносили на мембрану PVDF и блокировали в 5% обезжиренном молоке в течение 1 часа при комнатной температуре. Мембраны инкубировали в первичном антителе в течение ночи при 4°C, а затем добавляли вторичное антитело в течение 60 минут при комнатной температуре. MCF12F обрабатывали цисплатином и амигдалином по отдельности или в комбинации в течение 48 часов. Белки экстрагировали и разделяли с помощью электрофореза в 10%-ном полиакриламидном геле с додецилсульфатом натрия и зондировали с помощью антител против Bcl-2, фосфо-p53, p53, Bax, PUMA, PARP-1 и каспазы-9. Bcl-2 (CST 15071, 1:1000), PARP-1 (CST 9542, 1:1000), Bax (CST 2772, 1:1000), каспаза-9 (CTS 9502, 1:1000), фосфо-p53 (CST 9286 (Ser15), 1:1000), p53 (CST 9282, 1:1000), PUMA (CST 12450, 1:1000) были приобретены для Cell Signalling Technology (Дэнверс, Массачусетс, США), а Bcl-2 (sc-783, 1:1000), GAPDH (sc-25778, 1:1000) были приобретены у Santa Cruz Biotechnology Inc. Мы визуализировали полосы белков с использованием субстрата вестерн-блоттинга с улучшенной хемилюминесценцией (ECL) (Thermo Fisher Scientific) и машины Chemidoc от Biorad. Значения интенсивности из денситометрического анализа вестерн-блотов были нормализованы относительно GAPDH или β -актина с использованием программного обеспечения для анализа ImageJ. Значения интенсивности были выражены как кратное изменение по сравнению с контролем ( Ngoka, 2008 ).
Оценка апоптоза/некроза методом проточной цитометрии
Исследование индукции апоптоза/некроза оценивалось с помощью проточного цитометрического анализа при двойном окрашивании Annexin-V-FITC/пропидиум йодидом (PI). Клетки высевали в концентрации 1 × 10 5 клеток на лунку в 6-луночные планшеты для культивирования тканей и обрабатывали цисплатином (15 мкМ, 20 мкМ, 30 мкМ) и/или амигдалином (10 мМ), как указано. Клетки, обработанные только цисплатином, использовали в качестве положительного контроля, а необработанные клетки — в качестве отрицательного контроля. Клетки собирали и окрашивали с помощью набора Annexin V/Dead Cell Apoptosis (Life Technologies, Великобритания) в соответствии с инструкциями производителя. Апоптоз и некроз клеток анализировали с помощью проточного цитометра Attune NxT (Thermo Fisher Scientific, Великобритания) и программного обеспечения FlowJo v10 (BD Biosciences, США) ( Steensma et al., 2003 ).
Статистический анализ
Все результаты были представлены как среднее значение ± стандартная ошибка между самой низкой и самой высокой точками измерения. Непарные t -тесты были применены для исследования возможных различий в непрерывных переменных для двух групп. p -значения представлены как двусторонние с доверительными интервалами 95%. Статистический тест и анализ были проведены с использованием программного обеспечения Prism версии 5.0 (GraphPad, Сан-Диего, Калифорния, США). Для взаимодействия лекарств использовался метод Chou-Talalay (индекс комбинации) для оценки эффекта комбинированного лечения на основе данных о концентрации-эффекте ( Chou, 2010 ). Этот метод для комбинации лекарств основан на уравнении медианы-эффекта, которое исходит из принципа закона действия масс, который связывает один объект и несколько объектов, а также динамику первого и более высокого порядка ( Chou, 2011 ). Для результатов программного обеспечения уравнения индекса комбинации используется программа CompuSyn ( Chou, 2010 ).
Результаты
Оценка жизнеспособности нормальных и раковых клеток молочной железы после однократного лечения цисплатином или амигдалином
Чтобы определить влияние лечения амигдалином и цисплатином на нормальные (MCF12F) и раковые (MCF7, MDA-MB-231) клетки молочной железы, а также на фибробласты (FBS), мы использовали анализ MTT для оценки жизнеспособности клеток в зависимости от дозы в различные моменты времени. Во-первых, влияние амигдалина и цисплатина оценивалось по отдельности. Когда была указана идеальная концентрация (цисплатин 15 мкМ, амигдалин 10 мМ) для обоих видов лечения, была проведена комбинированная терапия с использованием цисплатина и амигдалина вместе для оценки разницы в количестве клеток.
Клетки MCF12F, MCF7, MDA-MB-231 и FBS обрабатывали 1, 10, 15, 20 и 30 мкМ цисплатина, а жизнеспособность клеток оценивали через 24, 48 и 72 ч ( рисунок 1A ). Цисплатин повлиял на все клеточные линии, уменьшив количество жизнеспособных клеток. Клетки рака молочной железы были поражены цисплатином больше, чем нормальные клетки молочной железы, а также фибробласты. Однако обе клеточные линии были значительно затронуты цисплатином.
РИСУНОК 1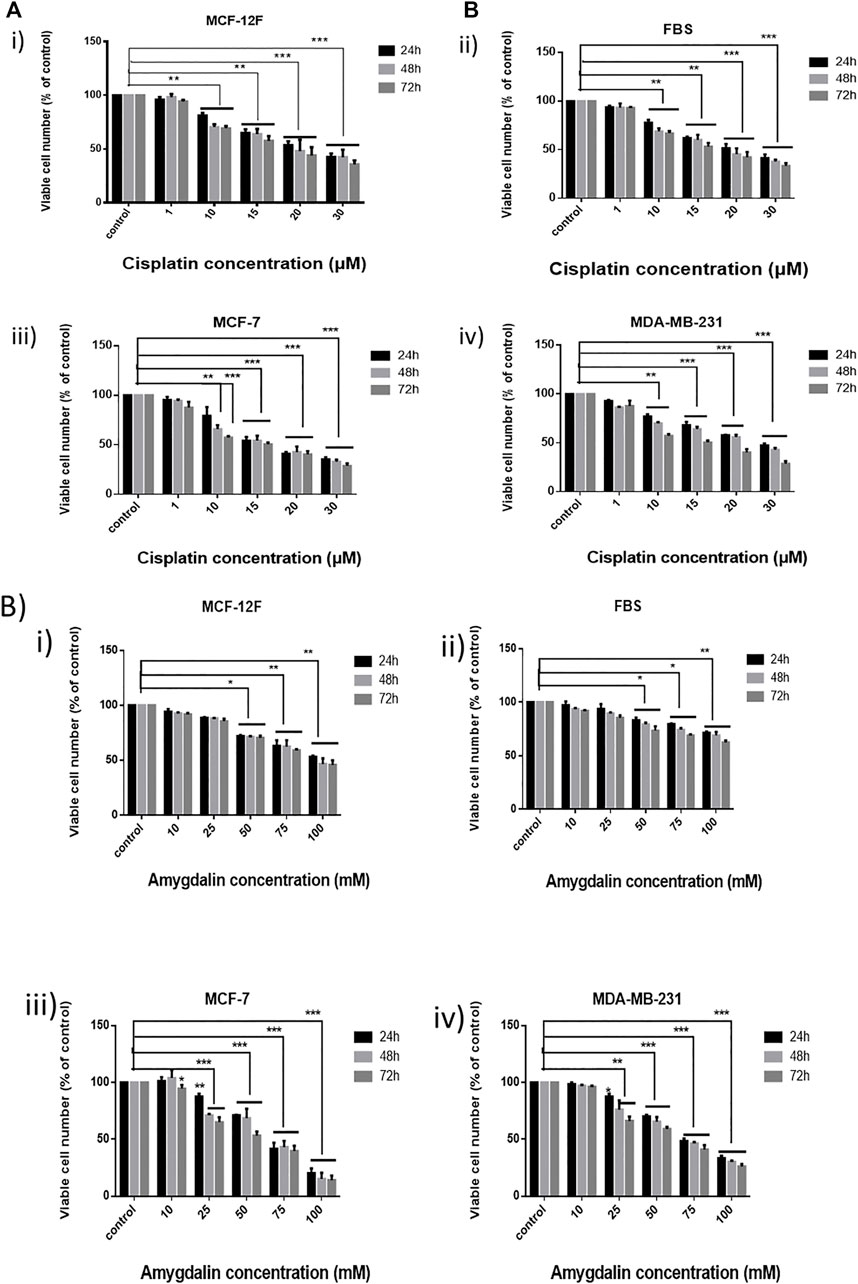
РИСУНОК 1. Эффект цисплатина и амигдалина в линиях клеток молочной железы, а также в фибробластах. (A) Анализ МТТ использовался для оценки цитотоксичности (% жизнеспособности клеток) возрастающих концентраций цисплатина (1, 10, 15, 20 и 30 мкМ) в i) MCF-12F, ii) FBS, iii) MCF-7 и iv) MDA-MB-231 в течение 24, 48, 72 ч и (B) возрастающих концентраций амигдалина (10, 25, 50, 75 и 100 мМ) в i) MCF-12F, ii) FBS, iii) MCF-7 и iv) MDA-MB-231 в течение 24, 48, 72 ч лечения. Звездочки указывают на силу статистической значимости между столбцами [* (0,05) < ** (0,01) < ***(0,001)].
Кроме того, клетки MCF12F, MCF7, MDA-MB-231 и FBS обрабатывали 10, 25, 50, 75 и 100 мМ амигдалина в течение 24, 48 и 72 ч ( Рисунок 1B ). Во всех клеточных линиях количество жизнеспособных клеток уменьшалось с увеличением концентрации амигдалина. Однако при использовании 10 мМ обработки амигдалином существенных различий не наблюдалось, поскольку уровень жизнеспособности клеток составлял >90% во всех временных точках. IC 50 цисплатина был определен равным 23,8 мкМ в MCF12F и 21,7 мкМ в MCF7, тогда как IC 50 амигдалина был равен 85,4 мМ в MCF12F и 64,5 мМ в MCF7 после 24, 48 и 72 ч обработки соответственно ( таблица 1 ).
Комбинированное лечение демонстрирует химиопротекторный эффект в нормальных клетках молочной железы
Комбинированное лечение применялось к клеткам MCF12F с использованием 10 мМ амигдалина и 1, 10, 15, 20 и 30 мкМ цисплатина. Сначала клетки обрабатывали амигдалином в течение 24 ч, а затем добавляли цисплатин на 24, 48 и 72 ч ( рисунки 2Ai–iii ). Затем мы предварительно обработали клетки 10 мМ амигдалином в течение 24 ч и наиболее подходящей концентрацией цисплатина, 15 мкМ в течение 24, 48 и 72 ч. Концентрация цисплатина основывалась на жизнеспособности клеток, которая составляла >50% при лечении одним агентом. Результаты показывают, что комбинированное лечение повышает жизнеспособность клеток во всех временных точках по сравнению с лечением одним цисплатином ( рисунок 2B ). Наиболее значительное увеличение жизнеспособности клеток ( ∼ 22%) наблюдалось через 48 ч при комбинированном лечении (24 ч амигдалин + 24 ч цисплатин). Анализ индекса комбинации, указывающий на синергизм или антагонизм амигдалина с цисплатином, представлен в таблице 1 .
РИСУНОК 2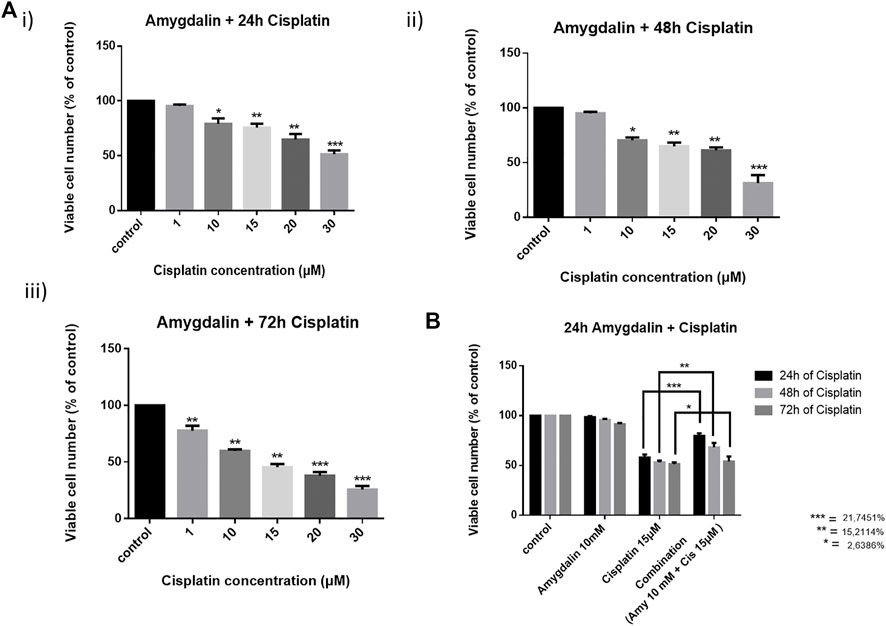
РИСУНОК 2. Эффект цисплатина отдельно и в сочетании с амигдалином в нормальных клетках молочной железы, MCF12F. Клетки были предварительно обработаны 10 мМ амигдалином, а затем цисплатин (1, 10, 15, 20 и 30 мкМ) был добавлен в течение (A) i) 24 ч, ii) 48 ч и iii) 72 ч. Анализ МТТ был применен для измерения жизнеспособности клеток в этих условиях. (B) Амигдалин (10 мМ) снижает цитотоксичность цисплатина (15 мкМ) и увеличивает выживаемость во всех временных точках с пиком на 48 ч общей обработки. Результаты представляют собой среднее значение ± SEM трех различных повторов и являются репрезентативными по крайней мере для трех различных экспериментов [* (0,05) < ** (0,01) < ***(0,001)].
Оценка апоптоза, опосредованного комбинированным лечением
Окрашивание аннексином V/PI использовалось для оценки апоптотического эффекта возрастающих концентраций цисплатина 15 и 20 мкМ с 10 мМ амигдалином или без него в течение 48 часов лечения.
Как показано на рисунке 3 , в клетках MCF12F процент поздних апоптотических клеток (двойной положительный результат по аннексину-V/PI) после 48-часовой комбинированной обработки (% среднего ± SE; цисплатин 15 мкМ + 10 мМ амигдалин: 5,8 ± 1,2 или цисплатин 20 мкМ + 10 мМ амигдалин: 4,3 ± 0,6) был значительно ниже по сравнению с обработкой цисплатином (15 мкМ: 9,5 ± 1,8; кратность изменения снижения = 1,6 или 20 мкМ: 19,3 ± 2,4; кратность изменения снижения = 4,5). Более того, % живых клеток при комбинированном лечении был увеличен (цисплатин 15 мкМ + 10 мМ амигдалин: 90,1 ± 1,2 или цисплатин 20 мкМ + 10 мМ амигдалин: 90,2 ± 0,5) по сравнению с применением только цисплатина (15 мкМ: 86,8 ± 0,5; p = 0,05 или 20 мкМ: 74,4 ± 1,9; p = 0,03 соответственно). Процент живых, ранних, поздних апоптотических и некротических клеток для всех условий лечения представлен на рисунке 3B . Была использована концентрация 30 мкМ, поскольку было обнаружено, что это самая высокая концентрация, способная убить более 50% обработанных клеток. Кроме того, 30 мкМ использовали в качестве положительного контроля для остальных экспериментов.
РИСУНОК 3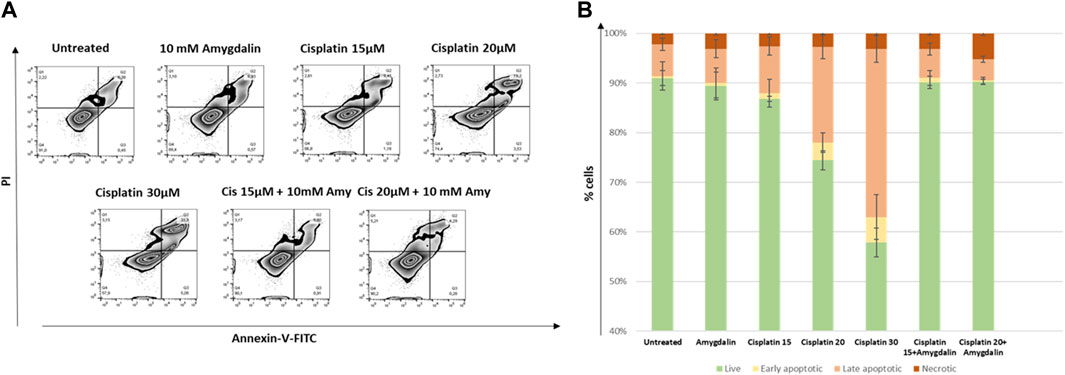
РИСУНОК 3. Влияние амигдалина на апоптоз, вызванный цисплатином в клетках MCF-12F. (A) Окрашивание аннексином V/PI использовалось для оценки апоптотического эффекта (% по сравнению с контролем) 15 и 20 мкМ цисплатина с 10 мМ амигдалином или без него в течение 48 ч. Приведены диаграммы зебра % живых (Q4), ранних (Q3), поздних апоптотических (Q2) и некротических клеток (Q1) при каждой стимуляции. (B) Сложенные столбчатые диаграммы, изображающие изменения в вышеупомянутых популяциях. Показаны данные трех независимых экспериментов.
Амигдалин и цисплатин регулируют белки, связанные с апоптозом
Комбинация цисплатина и амигдалина снизила уровни проапоптотических BAX, фосфо-p53, p53 и PUMA, медиаторов апоптотических ответов ( Рисунок 4 ). Напротив, уровни антиапоптотического BCl-2, известного ингибитора процесса проницаемости митохондриальной наружной мембраны (MOMP), были повышены ( Рисунок 4A ). PARP-1, фермент репарации ДНК, который расщепляется во время апоптоза, не представил свою расщепленную форму во время комбинированного лечения ( Рисунок 4B ). Расщепленная каспаза-9, мишень проапоптотических белков, высвобождаемых из митохондрий, была обнаружена в клетках, обработанных цисплатином, но не в группе комбинированного лечения ( Рисунок 4C ). Расщепленная каспаза-9 является активной формой, которая расщепляется от прокаспазы-9 (полной длины), показывая инициацию апоптотического пути. Эти результаты показывают, что комбинированное лечение с амигдалином подавляло апоптоз по сравнению с лечением только цисплатином.
РИСУНОК 4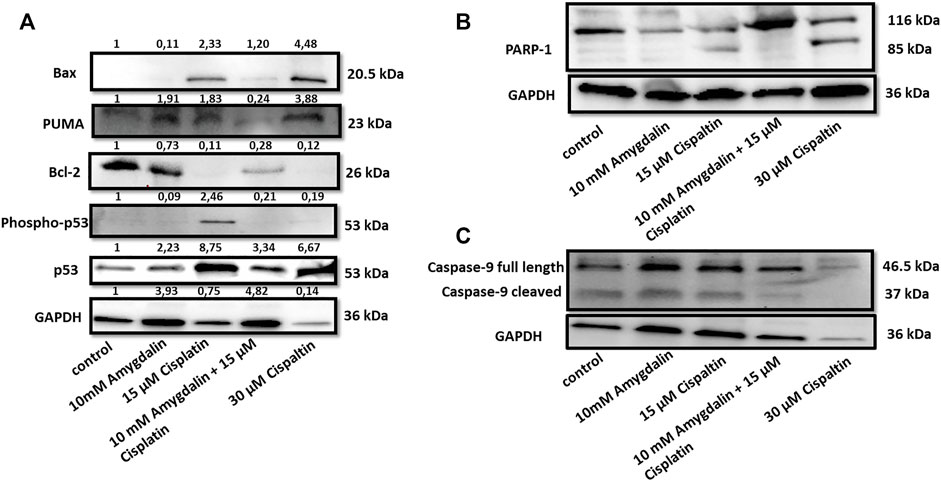
РИСУНОК 4. Влияние амигдалина и цисплатина на уровни и локализацию апоптотических белков в нормальных клетках. (A) Сочетание 10 мМ амигдалина и 15 мкМ цисплатина снизило уровни белков Bax, phosho-p53, p53, PUMA и увеличило уровни белков Bcl-2 после 48 ч обработки в MCF12F. (B) Показана экспрессия расщепленного PARP при обработке цисплатином. Цисплатин сам по себе вызывал расщепление каспазы-9, тогда как (C) амигдалин (10 мМ) снижал этот эффект. Значения интенсивности из денситометрического анализа вестерн-блотов показаны в верхней части каждого блота и были нормализованы по GAPDH с помощью программного обеспечения ImageJ. Результаты являются репрезентативными по крайней мере для трех независимых экспериментов.
Влияние амигдалина и цисплатина на экспрессию мРНК про- и антиапоптотических генов
После комбинированной обработки в течение 48 ч на клетках MCF12F уровни мРНК PUMA , p53 и Bax были значительно снижены на ∼83%, ∼66% и ∼44% соответственно по сравнению с обработкой только цисплатином. Напротив, уровни мРНК Bcl-2 и Bcl-xL были повышены на ∼44,5% и ∼51% соответственно при комбинированной обработке по сравнению с обработкой только цисплатином. Кроме того, уровни мРНК соотношения BAX/Bcl-2 были снижены на ∼81% при комбинированной обработке по сравнению с обработкой только цисплатином ( Рисунок 5 ). Более того, комбинированная обработка не показала никакого химиопротекторного эффекта на основе уровней экспрессии мРНК про- и антиапоптотических генов в клетках MCF7 и MDA-MB-231. ( Рисунок 5 ). Амигдалин сам по себе показал увеличение мРНК p53 в клетках MCF12F ( Рисунок 5 ), однако на уровне белка p53 ( Рисунок 4 ) он остался неизменным, и это может означать, что уровень мРНК не всегда коррелирует с уровнем белка. Аналогично, амигдалин не способствовал гибели клеток, как было оценено с помощью проточной цитометрии в клетках MCF12F ( Рисунок 3 ).
РИСУНОК 5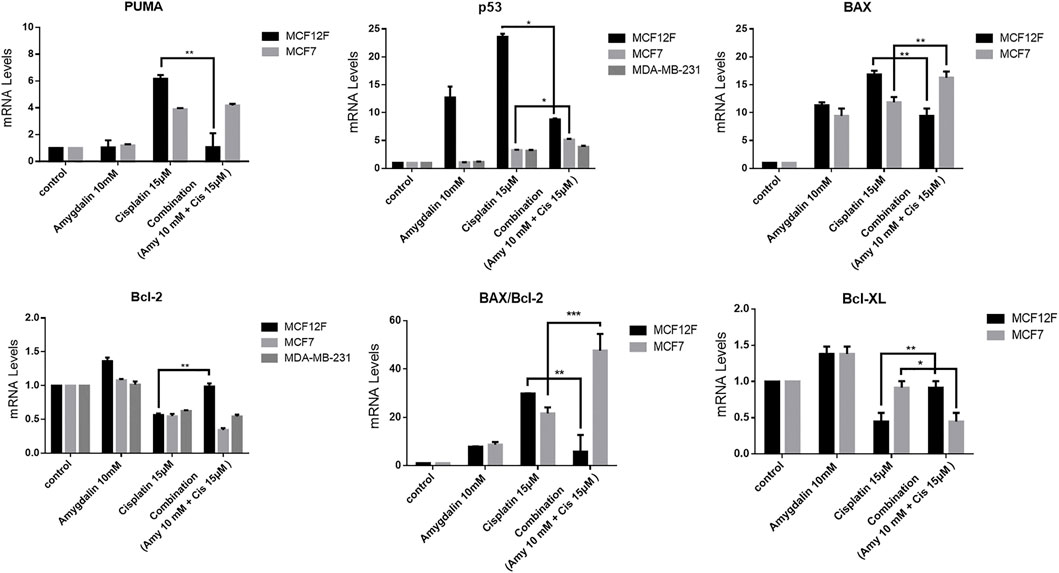
РИСУНОК 5. Влияние амигдалина и цисплатина на экспрессию мРНК проапоптотических и антиапоптотических генов. В MCF12F экспрессия мРНК проапоптотических генов PUMA, p53 и Bax , а также соотношение BAX/Bcl-2 были снижены при лечении комбинацией амигдалина (10 мМ) и цисплатина (15 мкМ) по сравнению с лечением цисплатином (15 мкМ), тогда как экспрессия мРНК антиапоптотических генов Bcl-2 и Bcl-xL была повышена при комбинированном лечении. В MCF7 экспрессия мРНК проапоптотических генов PUMA, p53 и Bax , а также соотношение BAX/Bcl-2 были увеличены при лечении комбинацией амигдалина (10 мМ) и цисплатина (15 мкМ) по сравнению с лечением цисплатином (15 мкМ), в то время как экспрессия мРНК антиапоптотических генов Bcl-2 и Bcl-xL была снижена при комбинированном лечении. В MDA-MB-231 экспрессия мРНК проапоптотического гена p 53 была увеличена при лечении комбинацией амигдалина (10 мМ) и цисплатина (15 мкМ) по сравнению с лечением цисплатином (15 мкМ), в то время как экспрессия мРНК антиапоптотического гена Bcl-2 была снижена. Результаты представляют собой среднее значение ± SEM трех различных повторов и являются репрезентативными по крайней мере для трех различных экспериментов, ∗ значение p < 0,05, ∗∗ значение p < 0,01, ∗∗∗ значение p < 0,001.
Обсуждение
Наиболее широко используемыми методами лечения рака являются хирургия, радиотерапия и химиотерапия, однако их эффективный терапевтический результат остается ограниченным из-за их неблагоприятных побочных эффектов, которые часто могут быть серьезными, что подчеркивает необходимость альтернативных или адъювантных методов лечения. Следовательно, стратегии, включающие фитохимические вещества, могут помочь в снижении этих побочных эффектов и улучшении качества жизни. Химиопротекторное лечение является многообещающим подходом, направленным на смягчение химиотерапевтических побочных эффектов в организме ( Maier et al., 2010 ). Фитохимические вещества, по-видимому, участвуют в профилактике и лечении рака из-за их относительно безопасного профиля цитотоксичности ( Duthie, 2007 ; Jagtap et al., 2009 ). В последнее десятилетие исследования направлены на выявление потенциала комбинированных стратегий с использованием одного или нескольких натуральных продуктов вместе с эффективным химиотерапевтическим средством для улучшения традиционной терапии рака ( Krzyzanowska et al., 2010 ; Kaminski et al., 2011 ; Saldanha and Tollefsbol, 2012 ).
В этом исследовании мы исследовали химиопротекторное и терапевтическое действие амигдалина в сочетании с обычным химиотерапевтическим средством цисплатином. Чтобы определить дифференциальную цитотоксичность по отношению к нормальным и раковым клеткам молочной железы, а также к фибробластам, эффект различных концентраций цисплатина и амигдалина, по отдельности, оценивался на клеточных линиях MCF12F, MCF7, MDA-MB-231, а также на фибробластах. Цисплатин и амигдалин снижали жизнеспособность клеток всех клеточных линий в зависимости от дозы и времени. Важно отметить, что обработка амигдалином в концентрации 10 мМ как нормальных, так и раковых клеток не показала статистически значимой разницы в жизнеспособности клеток, и этот показатель составлял более 90% во всех временных точках. Это подтверждает предыдущие выводы, указывающие на то, что многие фитохимические вещества, включая амигдалин, куркумин и сульфорафан, малотоксичны для нераковых, нормальных клеток ( Ravindran et al., 2009 ; Sharma et al., 2011 ).
На основании результатов IC50 концентрация амигдалина была установлена на уровне 10 мМ (не повлияла на жизнеспособность клеток ни в одной из клеточных линий в течение 24 ч). Концентрация цисплатина была установлена на уровне 15 мкМ для всех клеточных линий, чтобы иметь более 50% жизнеспособности клеток (для лучшей оценки эффекта комбинированного лечения в нормальных клетках).
Цисплатин является хорошо известным химиотерапевтическим препаратом для лечения широкого спектра злокачественных новообразований человека, но он связан с серьезными побочными эффектами и неспецифической цитотоксичностью, которая приводит к повреждению нормальных клеток и развитию лекарственной устойчивости ( McWhinney et al., 2009 ; Gopal et al., 2012 ). Предыдущие исследования продемонстрировали цитотоксический синергизм цисплатина и других агентов, таких как пчелиный яд, производное тиазоло[5,4-b] хинолина D3CLP и препарат AT-101 по отношению к различным линиям раковых клеток ( Alizadehnohi et al., 2012 ; Gonzalez-Sanchez et al., 2012 ; Mazumder et al., 2012 ; Karaca et al., 2013 ). Другое исследование продемонстрировало синергетическую активность фитохимического эпигаллокатехин галлата (EGCG) в сочетании с цисплатином и опухолеактивными соединениями палладия при раке яичников, что позволяет предположить, что комбинации платиновых препаратов, включая цисплатин и разработанные транс-палладии вместе с выбранными фитохимическими веществами, могут опосредовать преодоление лекарственной устойчивости в будущем. В нашем исследовании мы использовали комбинированную обработку 15 мкМ цисплатина и 10 мМ амигдалина как в нормальных клетках MCF12F, так и в FBS, чтобы дополнительно оценить безопасность амигдалина на нормальных клетках, а также его цитопротекторные способности в присутствии противораковых методов лечения. Также изучалась способность амигдалина сенсибилизировать эффекты, вызывающие смерть, в раковых клетках.
Проточная цитометрия подтвердила химиопротекторный эффект амигдалина, снижающий процент поздних апоптотических клеток до 4,5 раз ( рисунок 3 ). Как показано, амигдалин способен снижать цитотоксическое действие цисплатина на нормальные клетки молочной железы. Это подтверждается индексом комбинации (CI), который показал антагонизм между амигдалином и цисплатином при использовании в нормальных клетках и синергизм против раковых клеток.
Амигдалин приобрел широкую популярность благодаря своей противораковой активности и другим полезным эффектам на различные системы организма, таким как подавление фиброза почек, противоастматическое действие и улучшение иммунной функции ( Syrigos et al., 1998 ). Однако химиопротекторный потенциал амигдалина на нормальных эпителиальных клетках молочной железы ранее не исследовался.
Как показали наши результаты ( Рисунок 5 ), уровень мРНК проапоптотических PUMA, p53, BAX был значительно снижен, в то время как мРНК антиапоптотических Bcl-2 и Bcl-xL были повышены после комбинированного лечения по сравнению с лечением только цисплатином в нормальных клетках. Это конкретное наблюдение и открытие подтверждают основное обоснование механизма действия через апоптотический путь. Напротив, уровень мРНК проапоптотических PUMA, p53, BAX был повышен, в то время как мРНК антиапоптотических Bcl-2 и Bcl-xL были снижены после комбинированного лечения по сравнению с лечением только цисплатином в раковых клетках. Результаты ОТ-ПЦР в клетках рака молочной железы (MCF7 и MDA-MB-231) предполагают возможный синергический эффект амигдалина с цисплатином и подчеркивают его противораковую активность. Из предварительных отчетов известно, что возможным механизмом этого токсического эффекта является наличие β -глюкозидазы в раковых клетках, но не в нормальных клетках, которая способна расщеплять и высвобождать цианит из основной молекулы амигдалина. С другой стороны, нормальные клетки содержат фермент роданезу, который не может приводить к расщеплению и высвобождению цианида ( Newmark et al., 1981 ).
Похожая картина про- и антиапоптотической экспрессии также наблюдалась на уровнях белков. Экспрессия белков PUMA, p53, фосфо-p53 и Bax ( Рисунок 4 ) также снижалась при использовании комбинированного лечения по сравнению с одним цисплатином. С другой стороны, проапоптотические белки Bcl-2 и Bcl-xL демонстрировали повышенную тенденцию в присутствии амигдалина. Кроме того, при использовании комбинированного лечения были получены сниженные уровни расщепленной формы каспазы 9 и PARP, что указывает на ингибирование апоптотического пути. Мы поясняем, что даже несмотря на то, что можно наблюдать некоторую непоследовательность в отношении результата уровня GAPDH, учитывая полученный результат во всех других белках, запущенных для эксперимента по сравнению с GAPDH, непоследовательность нельзя считать проблемной. Более того, разница в расщеплении каспазы-9 между дорожкой 1 (контроль) и дорожкой 3 (15 мкМ цисплатин) очень слабая, но это обычное явление в ситуациях, когда наблюдается повышенный апоптотический результат.
Эти результаты могут указывать на то, что комбинированное лечение способно подавлять апоптоз по сравнению с лечением цисплатином.
Учитывая предыдущие изменения как в экспрессии мРНК, так и в экспрессии белка, мы можем сделать вывод, что комбинированное лечение цисплатином и амигдалином, возможно, может способствовать нормальному выживанию клеток выборочно, ингибируя апоптоз только в нормальных клетках. Эти наблюдения и выводы подтверждают нашу первичную и основную гипотезу и совместимы с другими исследованиями, которые указали на аналогичный механизм действия амигдалина в различных клеточных линиях или в моделях животных ( Kwon et al., 2003 ; Chang et al., 2006 ; Chen et al., 2013 ; Makarevic et al., 2014 ; Su et al., 2014 ).
В раковых клетках мы показали, что цисплатин способствует активации p53, что приводит к апоптозу посредством снижения уровня антиапоптотического белка Bcl-2 и повышения уровня проапоптотического белка Bax. Про- и антиапоптотические белки семейства Bcl-2 активируют процесс MOMP, который, в свою очередь, активирует апоптоз. MOMP приводит к высвобождению цитохрома c из митохондрий в цитозоль, вызывая активацию каспазы и последующий апоптоз ( Рисунок 6 ). С другой стороны, амигдалин оказывает цитопротекторное действие на нераковые клетки, способствуя эффективной экспрессии антиапоптотического гена.
РИСУНОК 6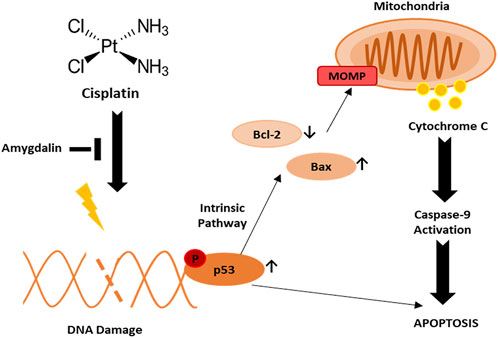
РИСУНОК 6. Потенциальный механизм действия амигдалина и цисплатина в нормальных клетках молочной железы против рака молочной железы. При лечении цисплатином индуцируется повреждение ДНК с последующей активацией фосфо-p53, приводящей к апоптозу. Фосфо-p53 может индуцировать апоптоз посредством снижения регуляции Bcl-2 и повышения регуляции Bax, что приводит к MOMP и активации каспазы-9.
Рак груди известен как одно из самых смертельных злокачественных новообразований среди женщин во всем мире и является серьезной проблемой общественного здравоохранения. Более того, известно, что существующие методы химиотерапии рака связаны с серьезными побочными эффектами, влияющими на качество жизни пациентов и продолжительность жизни. Таким образом, существует острая необходимость в новых терапевтических подходах. Это делает наши нынешние результаты важными для открытия новых горизонтов и направлений в области лечения рака.
Это исследование может быть расширено в будущем за счет использования экспериментов in vivo с использованием животных для оценки эффективности амигдалина при лечении рака. Предыдущие исследования показали, что они лечили мышей амигдалином с использованием 200, 100 и 50 мг/кг веса тела ( Albogami et al., 2020 ). Это правда, что используемые дозы концентрации (10–100 мМ) можно считать очень высокими для будущих исследований на животных. Тем не менее, тот факт, что они не были токсичными, дает нам уверенность в том, что они не будут способствовать возникновению побочных или токсических эффектов при использовании на животных. Кроме того, мы уточняем, что будут проведены новые исследования по оптимизации дозы, включая исследования ФК для обнаружения амигдалина в плазме перед использованием на животных.
Понимание цитопротекторного действия амигдалина во время химиотерапии может позволить разработать новые методы лечения, позволяющие снизить неблагоприятные побочные эффекты и, следовательно, улучшить качество и продолжительность жизни онкологических больных.
Заявление о доступности данных
Исходные данные, подтверждающие выводы настоящей статьи, будут предоставлены авторами без неоправданных оговорок.
Вклад автора
Задумал, разработал, контролировал и предоставил финансирование для исследования, IP, AS и PC; Обзор литературы и эксперименты, PC и PB; Проанализировал данные, PC, M-IC и CN; Сгенерировал и предоставил реагенты/материалы/инструменты анализа, CN и PP; Статистический анализ, PC и CN; Составил черновик рукописи: PC и IP; Отредактировал рукопись: AS и PP. Все авторы прочитали и согласились с опубликованной версией рукописи.
Благодарности
Авторы хотели бы выразить благодарность доценту Панайотису Политису за его любезную поддержку и предоставление использованных учебников.
Конфликт интересов
Авторы заявляют, что исследование проводилось при отсутствии каких-либо коммерческих или финансовых отношений, которые могли бы быть истолкованы как потенциальный конфликт интересов.
Примечание издателя
Все заявления, высказанные в этой статье, принадлежат исключительно авторам и не обязательно представляют заявления их аффилированных организаций или заявления издателя, редакторов и рецензентов. Любой продукт, который может быть оценен в этой статье, или заявление, которое может быть сделано его производителем, не гарантируется и не одобряется издателем.
Дополнительный материал
Дополнительный материал к этой статье можно найти в Интернете по адресу: https://www.frontiersin.org/articles/10.3389/fphar.2022.1013692/full#supplementary-material